Wie lange leben Kinder mit Melas-Syndrom? Seltene Krankheiten. Symptome des MELAS-Syndroms
Stichworte
MELAS-SYNDROM / MELAS-SYNDROM / EPILEPSIE / EPILEPSIE / KLINIKAnmerkung Wissenschaftlicher Artikel über klinische Medizin, Autor von wissenschaftlichen Arbeiten - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
Das MELAS-Syndrom ist eine genetisch bedingte Erkrankung aus der Gruppe der mitochondrialen Erkrankungen, definiert als mitochondriale Enzephalomyopathie mit Laktatazidose und Schlaganfall-ähnlichen Episoden (mitochondriale Enzephalomyopathie, Laktatazidose mit Schlaganfall-ähnlichen Episoden). Alle Organe und Gewebe sind am pathologischen Prozess beteiligt, aber das Muskel- und Nervensystem leidet stärker. Die Krankheit entwickelt sich am häufigsten im Alter zwischen 6 und 10 Jahren. Der Krankheitsverlauf ist progressiv. In den meisten Fällen manifestiert sich die Krankheit mit epileptischen Anfällen, wiederkehrenden Kopfschmerzen, Erbrechen und Anorexie. Epilepsie ist eine wichtige klinische Manifestation des MELAS-Syndroms. Epileptische Anfälle sind in 53 % der Fälle das erste erkennbare Symptom bei mitochondrialen Enzephalopathien (ME). Bei MELAS ist die okzipitale Epilepsie am häufigsten. Mit Fortschreiten der Erkrankung wird eine Therapieresistenz der Epilepsie festgestellt, oft mit Statusverlauf. Es sind Fälle der Umwandlung in die Koschewnikow-Epilepsie beschrieben. Wir präsentieren die Krankengeschichte eines Patienten mit einer Diagnose des MELAS-Syndroms, die zu Lebzeiten verifiziert wurde.
Verwandte Themen wissenschaftliche Arbeiten in der klinischen Medizin, Autor wissenschaftlicher Arbeiten - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
-
Mitochondriale Enzephalopathie mit schlaganfallartigen Episoden und Laktatazidose (Melas-Syndrom): Diagnostische Kriterien, Merkmale epileptischer Anfälle und Behandlungsansätze am Beispiel eines klinischen Falls
2017 / Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A. -
Schlaganfälle bei mitochondrialen Erkrankungen
2012 / Pizova N.V. -
Epilepsie bei Kindern mit mitochondrialen Erkrankungen: Merkmale der Diagnose und Behandlung
2012 / Zavadenko N. N., Kholin A. A. -
Neurologische Erkrankungen bei mitochondrialer Enzephalomyopathie – Laktatazidose mit schlaganfallähnlichen Episoden (MELAS-Syndrom)
2012 / Kharlamov Dmitry Alekseevich, Krapivkin Alexey Igorevich, Sukhorukov Vladimir Sergeevich, Kuftina Lyudmila Andreevna, Groznova Olga Sergeevna -
Melas-Syndrom als ungewöhnliche Ursache für Hypoparathyreoidismus: ein klinischer Fall
2018 / Umyarova Dilyara Shamilevna, Grebennikova Tatyana Alekseevna, Zenkova Tatyana Stanislavovna, Sorkina Ekaterina Leonidovna, Belaya Zhanna Evgenievna -
Schlaganfall-ähnliche Episoden bei mitochondrialer Enzephalomyopathie mit Laktatazidose
2010 / Kalashnikova Lyudmila Andreevna, Dobrynina L. A., Sakharova A. V., Chaikovskaya R. P., Mir-kasimov M. F., Konovalov R. N., Shabalina A. A., Kostyreva M. V., Gnezditsky V. V., Protsky S. V. -
Mitochondriale Zytopathien: Melas und MIDD-Syndrome. Ein genetischer Defekt, verschiedene klinische Phänotypen
2017 / Muranova A.V., Strokov I.A. -
Gutartige okzipitale Epilepsie des Kindesalters mit frühem Beginn (Panayotopoulos-Syndrom). Beschreibung des klinischen Falls
2015 / Matyuk Yu.V., Kotov A.S., Borisova M.N., Panteleeva M.V., Shatalin A.V. -
Polymorphismus der klinischen Manifestationen einer progressiven mitochondrialen Enzephalomyopathie im Zusammenhang mit einer POLG1-Genmutation
2016 / Yablonskaya M.I., Nikolaeva E.A., Shatalov P.A., Kharabadze M.N. -
Diagnostischer Wert der Untersuchung der zytochemischen Aktivität von Enzymen bei erblichen mitochondrialen Erkrankungen
2017 / Kazantseva I.A., Kotov S.V., Borodataya E.V., Sidorova O.P., Kotov A.S.
EPILEPSIE BEI MELAS-SYNDROM
Das MELAS-Syndrom ist eine genetisch bedingte Erkrankung der mitochondrialen Gruppe, definiert als mitochondriale Enzephalomyopathie, Laktatazidose mit schlaganfallähnlichen Episoden. Der pathologische Prozess betrifft alle Organe und Gewebe, ist aber meistens nachteilig für das Muskel- und Nervensystem. Die Erkrankung tritt am häufigsten bei Kindern im Alter von 6 bis 10 Jahren auf. Der klinische Verlauf ist progredient. In den meisten Fällen manifestiert sich die Krankheit durch epileptische Anfälle, rezidivierende Kopfschmerzen, Erbrechen, Anorexie. Das wichtige klinische Erscheinungsbild des MELAS-Syndroms ist Epilepsie. Epileptische Anfälle sind in 53 % der Fälle das erste Diagnosesymptom einer mitochondrialen Enzephalopathie (ME). Okzipitale Epilepsie ist die häufigste beim MELAS-Syndrom. Mit fortschreitender Krankheit wird eine Behandlungsresistenz der Epilepsie beobachtet, oft mit dem Auftreten eines Status epilepticus. Es werden einige Fälle der Transformation in die Kozhevnikov-Epilepsie beschrieben und die Anamnese eines Patienten mit einer zu Lebzeiten verifizierten MELAS-Syndrom-Diagnose angegeben.
Der Text der wissenschaftlichen Arbeit zum Thema "Epilepsie beim Melasyndrom"
BAND IV AUSGABE 3 2009
EPILEPSIE MIT MELAS-SYNDROM
K. Yu. Muchin1, M.B. Mironov1, N.V. Nikiforova1, C.B. Mikhailova2, VA. Chadaev1, AA. Alikhanov1-2, B.N. Ryzhkov1, A.S. Petruchin1
EPILEPSIE BEI MELAS-SYNDROM
KYu. Muchin1, M.B. Mironov1, N.V. Nikiforova1, S.V. Mikhailova2, UA. Chadaev1, AA. Alikhanov1-2, B.N. Ryzkov1 AS. Petruchin1
1 - Abteilung für Neurologie und Neurochirurgie, Fakultät für Pädiatrie, Staatliche Bildungseinrichtung für Höhere Berufsbildung, Russische Staatliche Medizinische Universität Roszdrav
2 - Klinisches Krankenhaus für russische Kinder
Das MELAS-Syndrom ist eine genetisch bedingte Erkrankung aus der Gruppe der mitochondrialen Erkrankungen, definiert als mitochondriale Enzephalomyopathie mit Laktatazidose und Schlaganfall-ähnlichen Episoden (mitochondriale Enzephalomyopathie, Milchsäure mit Schlaganfall-ähnlichen Episoden). Alle Organe und Gewebe sind am pathologischen Prozess beteiligt, aber das Muskel- und Nervensystem leidet stärker. Die Krankheit entwickelt sich am häufigsten im Alter zwischen 6 und 10 Jahren. Der Krankheitsverlauf ist progressiv. In den meisten Fällen manifestiert sich die Krankheit mit epileptischen Anfällen, wiederkehrenden Kopfschmerzen, Erbrechen und Anorexie. Epilepsie ist eine wichtige klinische Manifestation des MELAs-Syndroms. Epileptische Anfälle sind in 53 % der Fälle das erste erkennbare Symptom bei mitochondrialen Enzephalopathien (ME). Bei MELAS ist die okzipitale Epilepsie am häufigsten. Mit Fortschreiten der Erkrankung wird eine Therapieresistenz der Epilepsie festgestellt, oft mit Statusverlauf. Es sind Fälle der Umwandlung in die Koschewnikow-Epilepsie beschrieben. Wir präsentieren die Krankengeschichte eines Patienten mit einer Diagnose des MELAS-Syndroms, die zu Lebzeiten verifiziert wurde.
Schlüsselwörter: MELAS-Syndrom, Epilepsie, Klinik, Diagnostik, Behandlung.
Das MELAS-Syndrom ist eine genetisch bedingte Erkrankung der mitochondrialen Gruppe, definiert als mitochondriale Enzephalomyopathie, Laktatazidose mit schlaganfallähnlichen Episoden. Der pathologische Prozess betrifft alle Organe und Gewebe, ist aber meistens nachteilig für das Muskel- und Nervensystem. Die Erkrankung tritt am häufigsten bei Kindern im Alter von 6 bis 10 Jahren auf. Der klinische Verlauf ist progredient. In den meisten Fällen manifestiert sich die Krankheit durch epileptische Anfälle, rezidivierende Kopfschmerzen, Erbrechen, Anorexie. Das wichtige klinische Erscheinungsbild des MELAS-Syndroms ist Epilepsie. Epileptische Anfälle sind in 53 % der Fälle das erste Diagnosesymptom einer mitochondrialen Enzephalopathie (ME). Okzipitale Epilepsie ist die häufigste beim MELAS-Syndrom. Mit fortschreitender Krankheit wird eine Behandlungsresistenz der Epilepsie beobachtet, oft mit dem Auftreten eines Status epilepticus. Es werden einige Fälle der Transformation in die Kozhevnikov-Epilepsie beschrieben und die Anamnese eines Patienten mit einer zu Lebzeiten verifizierten MELAS-Syndrom-Diagnose angegeben.
Schlüsselwörter: MELAS-Syndrom, Epilepsie, Krankheitsbild, Diagnostik, Behandlung.
Das MELAS-Syndrom ist eine genetisch bedingte Erkrankung aus der Gruppe der mitochondrialen Erkrankungen, definiert als mitochondriale Enzephalomyopathie mit Laktatazidose und schlaganfallähnlichen Episoden (mitochondriale Enzephalomyopathie, Laktatazidose mit schlaganfallähnlichen Episoden).
Das MELAS-Syndrom wurde erstmals als eigenständige nosologische Form von S. Pavlakis et al. im Jahr 1984 . Einige Autoren schlagen jedoch vor, dass die Krankheit früher unter dem Namen "familiäre Polyodystrophie, mitochondriale Myopathie, Laktatazidämie" beschrieben wurde.
Die Prävalenz in der Bevölkerung wurde nicht ermittelt. Bis zum Jahr 2000 wurden mehr als 120 Beobachtungen des MELAS-Syndroms veröffentlicht, auch in der heimischen Presse.
Das MELAS-Syndrom wird in 25 % der Fälle mit hohem Risiko maternal vererbt, bei 56-75 % der Patienten ist die Familienanamnese jedoch nicht belastet. Die Krankheit ist mit Mutationen in mitochondrialen DNA-Genen verbunden, die Untereinheiten von Atmungskettenkomplexen und Transport-RNA-Genen (MT-ND1, MT-ND5, MT-TH, MT-TL1 und MT-TV) codieren. In 80-90 % der Fälle des MELAS-Syndroms beruht die Krankheit auf einer Punktmutation im MT-TL1-Gen, das für die Leucin-Transfer-RNA kodiert. Bei dieser Mutation wird das Adenin-Nukleotid an Position 3243 (A3243G) durch Guanin ersetzt, was die Synthese aller Proteine in den Mitochondrien stört.
Alle Organe und Gewebe sind am pathologischen Prozess beteiligt, aber das Muskel- und Nervensystem leidet stärker.
Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova C.V., Chadaev V.A., Alikhanov A.A., Ryzhkov BN., Petrukhin A.S.
Epilepsie bei MELAS-Syndrom Rus. zhur. det. Neur.: Bd. IV, No. 3, 2009.
ORIGINALARTIKEL
Themen als die volatilsten. Die Schwere der klinischen Manifestationen hängt vom Schwelleneffekt (Alter, Energiebedarf der Gewebe), von der Kontrolle der Kerngene über die Synthese der Atmungskette, Heteroplasmie (unterschiedlicher Gehalt an mutierten mtDNA-Molekülen im Gewebe) ab. Es wurde gezeigt, dass bei Patienten mit dem MELAS-Syndrom der Gehalt an mutierter mtDNA in verschiedenen Geweben 93-96 % beträgt. Bei Familienmitgliedern von Probanden wird auch mutierte mtDNA in den Geweben nachgewiesen, ihr Gehalt ist jedoch deutlich geringer: 62-89% in der gelöschten Form der Krankheit, von 28 bis 89% ohne klinische Anzeichen des Syndroms.
Die Krankheit entwickelt sich am häufigsten im Alter von 6 bis 10 Jahren, aber es gibt Fälle von einem früheren (bis zu zwei Jahren) oder späteren Debüt - von 21 bis 40 Jahren. Vor dem Ausbruch der Krankheit entwickeln sich 90-100% der Patienten normal. Der Krankheitsverlauf ist progressiv, bösartiger mit frühem Beginn.
In den meisten Fällen manifestiert sich die Krankheit mit epileptischen Anfällen, wiederkehrenden Kopfschmerzen, Erbrechen und Anorexie. Sie sollten auch auf Intoleranz gegenüber körperlicher Aktivität in Form von Verschlechterung des Gesundheitszustands und Auftreten von Muskelschwäche achten. Der myopathische Symptomkomplex äußert sich in Belastungsintoleranz, Muskelschwäche, Müdigkeit und manchmal Muskelhyptrophie.
Im weiteren Verlauf entwickelt sich meist eine Demenz. Symptome wie zerebelläre Ataxie, neurosensorische Taubheit und periphere Polyneuropathie sind seltener.
Charakteristisch sind schlaganfallartige Episoden, die sich durch rezidivierende Kopfschmerzattacken, Schwindel, die Entwicklung fokaler neurologischer Symptome (Paresen, Hemianopsie) und Koma äußern können. Diese akuten Episoden werden oft durch Fieber oder interkurrente Infektionen ausgelöst. Diese Manifestationen können eine ziemlich schnelle Regression (von mehreren Stunden bis zu mehreren Wochen) sowie eine Rückfallneigung aufweisen.
Epilepsie ist eine wichtige klinische Manifestation, die häufig in den frühen Stadien von MELAS auftritt. Das
oft die offensichtlichste neurologische Manifestation, insbesondere bei der atypischen mitochondrialen Enzephalopathie (ME). Epileptische Anfälle sind in 53 % der Fälle das erste erkennbare Symptom bei mitochondrialen Enzephalopathien (ME).
Bei MELAS ist die okzipitale Epilepsie (SE) am häufigsten. Charakterisiert durch fokale Anfälle mit Ursprung in den Okzipitallappen. Krampfanfälle sind oft mit vorübergehenden oder anhaltenden neurologischen Symptomen wie Gesichtsfeldverlust verbunden.
Anfälle, die von der Okzipitalrinde ausgehen, werden nach ihrer Manifestation in subjektive Empfindungen (Aura) und in klinisch nachweisbare Symptome, in der Regel mit einer motorischen Komponente, eingeteilt. Epileptische Auren, die vom Okzipitallappen ausgehen, umfassen einfache und komplexe visuelle Halluzinationen, Amaurose. Die typischsten für SE charakteristischen Anfälle sind einfache visuelle Halluzinationen, die sich als positive (Blitze, Lichtflecke) und negative Symptome (Skotom, Hemianopsie) äußern können. Am häufigsten werden visuelle Halluzinationen als ein Punkt oder Lichtpunkte beschrieben, die entweder konstant oder blinkend sind. In der Regel ist der Fleck weiß mit einem grünlichen Farbton. Halluzinationen können auch mehrfarbig oder einfarbig sein. Halluzinationen treten meist kontralateral zum Erregungsherd im okzipitalen Cortex in den Gesichtsfeldern mit anschließender Ausbreitung auf. Es sollte jedoch beachtet werden, dass in den Beschwerden von Patienten die visuelle Aura nicht oft erkannt wird.
Komplexe visuelle Halluzinationen werden beobachtet, wenn sich die epileptische Erregung auf die okzipito-temporalen oder okzipito-parietalen Regionen ausbreitet. Komplexe visuelle Halluzinationen können in Form von Menschen, Tierobjekten oder Szenen auftreten, vertraut oder ungewohnt, angenehm oder erschreckend, beängstigend, einfach oder grotesk sein, können statisch sein oder sich in einer horizontalen Ebene bewegen und verschwinden. In der Regel sind sie ein Endsymptom vor der Entwicklung eines motorischen Anfalls; kann das erste iktale Symptom sein, tritt aber häufiger danach auf
BAND IV AUSGABE 3 2009
grundlegende Halluzinationen.
Die iktale Ama vrosis ist eine spezielle, extrem schwer zu diagnostizierende Art von Anfällen, die vom okzipitalen Cortex ausgehen. Nach Ansicht vieler Autoren ist dies das gleiche häufige Symptom einer Reizung des Okzipitallappens sowie visueller Halluzinationen, bleibt jedoch oft unerkannt. Normalerweise unterscheiden Patienten dieses Symptom nicht gesondert in der Struktur des Angriffs. Der Sehverlust tritt bilateral mit Verlust der lateralen Felder auf. Mögliche homonyme Hemianopsie kontralateral zum Anfallsherd. Die Empfindungen der Patienten werden von ihnen als dunkler werdende Augen, „weiße Dunkelheit“, beeinträchtigte Farbwahrnehmung beschrieben. Vielleicht ein Statusverlauf mit der Ausbildung des sogenannten Status epilepticus amamauroticus.
Okzipitale Anfälle können mit autonomen Symptomen einhergehen. Dazu gehören Migräne, Schwindel, Übelkeit und Erbrechen. Ein häufiges Symptom sind migräneartige Kopfschmerzen nach dem Angriff.
Die klinischen Manifestationen von Anfällen, die begrenzt im Okzipitalkortex auftreten, sind durch eine Abweichung der Augen zur Seite gekennzeichnet. Die Abweichung der Augen kann zusammen mit der Abweichung des Kopfes zur Seite festgestellt werden. In den meisten Fällen wird eine Abweichung der Augen zum kontralateralen Fokus festgestellt. Es werden jedoch Fälle beschrieben, in denen eine Abduktion der Augen in Richtung des Fokus beobachtet wird. Eines der Merkmale von "okzipitalen" Anfällen ist auch die sofortige Verteilung der Entladung auf die vorderen Teile des Gehirns, während das klinische Bild in der Regel von einer ausgeprägten motorischen Komponente dominiert wird. Tonische, tonisch-klonische (sowohl hemkonvulsive als auch sekundär generalisierte), automotorische Anfälle sind möglich. In diesem Zusammenhang ist es wichtig, die anfänglichen klinischen Symptome zu identifizieren - ein unmotiviertes und plötzliches Stoppen des Blicks, das Betrachten nicht vorhandener Objekte, ein unvernünftiges Lächeln, vegetative Manifestationen und die notwendige Dokumentation der primären iktogenen Zone mit der VEM-Methode.
Mit Fortschreiten der Erkrankung wird eine Therapieresistenz der Epilepsie festgestellt, oft mit Statusverlauf. Es werden Fälle der Transformation in eine Kozhevnikov-Epilepsie beschrieben. Eine Reihe von Auto-
Rov beschreibt die Möglichkeit eines Status epilepticus als erstes Symptom bei Patienten mit MELAS ohne vorherige epileptische Anfälle in der Vorgeschichte. Ribacoba R. et al. beschreiben in ihrer Veröffentlichung 4 Fälle der Entwicklung einer Epilepsie partialis continua mit fokalen motorischen Anfällen, denen eine Anamnese von Episoden von Migränekopfschmerzen vorausging. Miyazaki M. et al. zeigten die Möglichkeit eines fortgesetzten fokalen Myoklonus innerhalb einer Epilepsie partialis continua bei Patienten mit MELAS. Araki T. et al. beobachtete einen Patienten im Alter von 37 Jahren mit epileptischem Status von fokalen Anfällen in Form von Bewusstseinsschwankungen, homonymer Hemianopsie in Kombination mit paroxysmalen Episoden von Augenabweichungen zur Seite. Das EEG zeichnete fortgesetzte EEG-Muster von Anfällen auf, die in der Okzipitalregion lokalisiert waren. Bei erwachsenen Patienten mit MELAS überwiegen fokale motorische Anfälle, das EEG zeigt jedoch eine Dominanz multiregionaler epileptiformer Aktivität in den Okzipitalregionen.
Epileptiforme Aktivität wird in 71 % der Fälle nach Beginn der Anfälle registriert. Eine elektroenzephalographische Studie an Patienten mit MELAS-Syndrom ist durch epileptiforme Aktivität in den Okzipitalregionen gekennzeichnet. Eine Reihe von Autoren assoziieren das Auftreten regionaler epileptiformer Störungen mit Schlaganfällen. Laut der Studie von Fujimoto S. hatte die Mehrheit der untersuchten Patienten mit MELAS-Syndrom in der Akutphase (d. h. innerhalb von 5 Tagen nach einer schlaganfallähnlichen Episode) regionale Delta-Wellen mit hoher Amplitude in Kombination mit Polyspikes. Die Autoren schlagen vor, dieses Muster als pathognomonisch für schlaganfallähnliche Episoden zu betrachten. Die epileptiforme Aktivität kann sich neben den Okzipitalregionen auf die Schläfenregionen, bifrontal und bei diffuser Verteilung auch bilateral auf die hinteren Regionen ausbreiten. Möglicherweise das Auftreten einer photoparoxysmalen Reaktion während der rhythmischen Photostimulation.
Das führende Laborzeichen ist ein Anstieg des Laktatspiegels im Blut.
ORIGINALARTIKEL
wi über 2,0 mmol / l, was zur Entwicklung einer Laktatazidose führt.
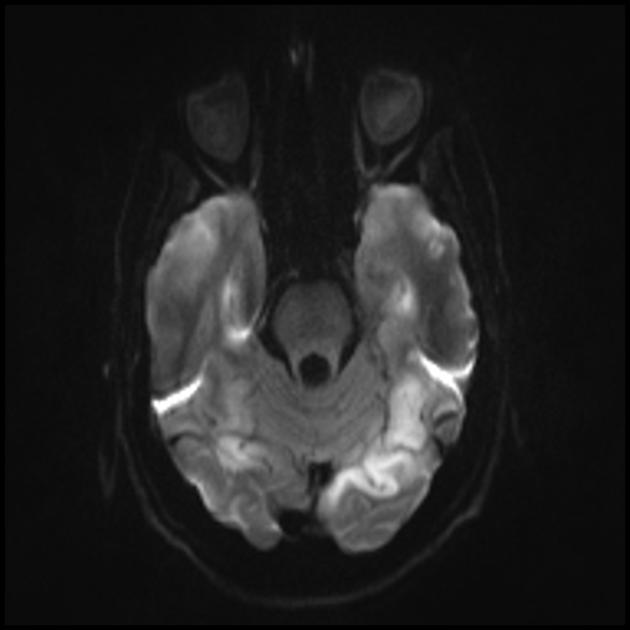
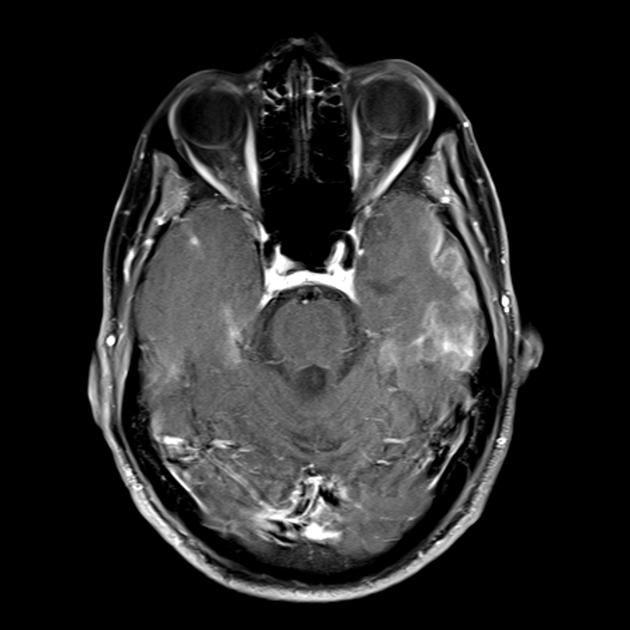
Ein MRT des Gehirns im Frühstadium der Erkrankung kann unauffällig sein, auch wenn eine Epilepsie auftritt. Bildgebende Verfahren zeigen Infarktzonen in den Großhirnhemisphären (80 %), seltener im Kleinhirn und in den Basalganglien. Es kann auch zu einer Verkalkung der Basalganglien, einer Atrophie der Großhirnrinde kommen. In einer Photonenemissionsstudie wird die Akkumulation des Isotops 3-16 Tage vor dem Auftreten der Infarktzone (Abnahme des Isotopensignals) auf einem Computertomogramm des Gehirns nachgewiesen. Die MRT des Gehirns zeigt Läsionen, die sich vorwiegend in den Hinterhauptslappen befinden und vorübergehend sein können. Der Okzipitalkortex ist überwiegend betroffen, die weiße Substanz ist weniger geschädigt. Auf T2-gewichteten Bildern erscheinen Hirnläsionen bei MELA als Bereiche mit erhöhter Signalintensität. Vorübergehende hyperintense Areale werden von einer Reihe von Autoren mit reversiblen Gefäßödemen in Verbindung gebracht.
Die Angiographie zeigt normalerweise keine vaskulären Anomalien. Die diffusionsgewichtete MRT zeigt Veränderungen im Zusammenhang mit vasogenen Ödemen.
Histopathologie: Muskelbiopsie zeigt Fasern mit zerrissenen „roten Rändern“. Die Autopsie des Gehirns ist gekennzeichnet durch eine Kombination aus alten und neuen Infarktherden sowie einer Atrophie des Kortex mit fokalen Nekroseherden.
Derzeit ist die Therapie unterstützend. Die Hauptrichtung der Behandlung ist die Verbesserung der Energiebilanz der Mitochondrien und der Atmungskette. Wenden Sie Coenzym p10 (80–300 mg/Tag), Vitamine K1 und KZ (25 mg/Tag), Bernsteinsäure (bis zu 6 g/Tag), Vitamin C (2–4 g/Tag), Riboflavin (100 mg/Tag) an. Tag) und Nicotinamid (bis zu 1 g/Tag). Im Zusammenhang mit dem sich entwickelnden sekundären Carnitinmangel wird den Patienten L-Carnitin (bis zu 100 mg/kg/Tag) verschrieben. Vitamin E (300-500 mg/Tag) und Vitamin C (2-4 mg/Tag) werden als antioxidative Therapie eingesetzt.
Es gibt keine allgemein akzeptierten antiepileptischen Therapieschemata für MELA. Einige Autoren schlagen vor, Medikamente auszuschließen, die den Energiestoffwechsel hemmen können (Barbiturate, Valproinsäure-Medikamente sowie einige Medikamente aus anderen Gruppen, z. B. Chloramphenicol). In der Literatur sind mehrere Einzelfälle von Anfallsverschlimmerung unter Anwendung von Valproinsäure beim MELA-Syndrom mit der A3243C-Mutation beschrieben. Die wichtigsten Antiepileptika bei der Behandlung von Epilepsie beim MELA-Syndrom sind Tegretol (oder Trileptal), Topamax, Keppra in durchschnittlichen therapeutischen Dosen. Eine richtig ausgewählte Therapie führt zu einer signifikanten Abnahme der Häufigkeit von sekundären generalisierten Krampfanfällen. Anfälle mit beeinträchtigten vegetativ-viszeralen und visuellen Funktionen sind jedoch in der Regel therapieresistent. Im Endstadium der Erkrankung kann die Häufigkeit epileptischer Anfälle abnehmen.
Hier ist die Krankengeschichte eines Patienten mit einer Diagnose des MELAY-Syndroms, die zu Lebzeiten verifiziert wurde.
Patient Ch.A., 11 Jahre alt, wurde im Zentrum für pädiatrische Neurologie und Epilepsie beobachtet. Bei der Aufnahme wurde über einen allmählichen Verlust der Sprachfähigkeit, eine ausgeprägte Gangstörung mit Gehverweigerung, eine deutliche Sehminderung, Launenhaftigkeit und negatives Verhalten geklagt; tägliche serielle Attacken in Form von Zuckungen der Gesichtsmuskeln, Muskeln der oberen und unteren Extremitäten sowie kurzfristige Episoden von Sehverlust.
Das Debüt der Krankheit wurde im Alter von 5 Jahren und 9 Monaten festgestellt. Zum ersten Mal traten vor dem Hintergrund der vollen Gesundheit beim Einschlafen starke Kopfschmerzen auf, einfache visuelle Halluzinationen ("gelber Strahl"), gefolgt von einer heftigen Drehung der Augen und des Kopfes zur Seite und der Entwicklung eines Generals tonisch-klonischer Krampfanfall, nach dem Erbrechen festgestellt wurde. Nach 9 Monaten Attacken mit den gleichen Symptomen wiederholten sich und nahmen schnell Seriencharakter an. Nach der Ernennung von Tegretol in einer Dosis von 400 mg pro Tag verringerte sich die Häufigkeit der Anfälle auf 1 Mal pro Monat. Tegretol wurde durch Depakine Chrono in einer Dosis von 900 mg/Tag ersetzt, wogegen eine klinische Remission für 6 Monate festgestellt wurde. In Anbetracht des klinischen Symptoms
BAND IV AUSGABE 3 2009
Tomatik, Beschränkung der Anfälle auf die Zeit des Einschlafens, normale Intelligenz des Patienten, positive Reaktion auf Valproat, idiopathische okzipitale Epilepsie wurde diagnostiziert.
Im Alter von 7 Jahren setzten die fokalen versiven Anfälle mit sekundärer Generalisierung beim Einschlafen mit der gleichen Häufigkeit von 1 Mal pro Monat fort. Eine Erhöhung der Dosis von Depakine auf 1500 mg/Tag führte nicht zu einer Verringerung der Anfallshäufigkeit. Wenn Lamiktal in einer Dosis von 75 mg/Tag hinzugefügt wurde, hörten die Attacken für 4 Monate auf und setzten dann mit der gleichen Häufigkeit wieder ein. Im Alter von 8 Jahren kamen Attacken mit kurzzeitigem Sehverlust hinzu. Ab 8 Jahre 8 Monate vor dem Einschlafen traten atypische Abwesenheiten auf: schnelles Blinzeln mit Schließen der Augenlider und Aufrichten der Augäpfel; Bewusstsein schwankt.
Im Alter von 9 Jahren traten mehrere mehrtägige Serienattacken mit einfachen visuellen Halluzinationen in Form eines aufblitzenden "Strahls" vor den Augen auf, mit einer Drehung der Augen und des Kopfes nach rechts. Vor dem Einschlafen verwandelten sich solche Attacken manchmal in fokale hemiklonische Attacken, die sich durch Reduktion des Gesichts manifestierten
Muskulatur rechts, Zucken des Kopfes nach rechts, Klonien der rechten Gliedmaßen (größer als der Arm). Manchmal gab es nach dem Anfall starke Kopfschmerzen und Erbrechen. Im gleichen Alter traten hemmende Anfälle auf: eine Aura in Form von Gänsehaut in der großen Zehe des rechten Fußes, gefolgt von einer kurzzeitigen Schwäche des rechten Beins und einer Unbeholfenheit der rechten Hand. Topamax wurde mit einer Dosis von 100 mg/Tag in das Behandlungsschema eingeführt – es gab 1 Jahr lang keine epileptischen Anfälle.
Außerdem traten im Alter von 9 Jahren erstmals paroxysmale Zustände auf, begleitet von starken Kopfschmerzen, Erbrechen und der Entwicklung einer rechtsseitigen Hemiparese. In einigen Fällen wurden solche Zustände von einer Amaurose begleitet, die mehrere Minuten bis mehrere Tage anhielt.
Im Alter von 10,5 Jahren traten die Attacken wieder auf in Form von Kopfdrehung nach links, ruckartige Bewegungen der Augäpfel nach links, Dauer bis zu 5 s, Häufigkeit bis zu 3 mal pro Stunde, täglich, auch im Schlaf. Die Topamax-Dosis wurde ohne signifikante Wirkung auf 150 mg/Tag erhöht. Mit 10 Jahren 10 Monaten. nach starken Kopfschmerzen, abwechselnd
Reis. 1. Patient Ch.A. 10 Jahre. Diagnose: MEAE-Syndrom. Symptomatische fokale Epilepsie.
Video-EEG-Monitoring (2004): Vor dem Hintergrund einer diffusen Verlangsamung der Hauptaktivität des Gehirns wurde eine fortgesetzte epileptiforme Aktivität in der linken Okzipitalregion registriert. Subklinische EEG-Muster eines Anfalls wurden auch in der linken Okzipitalregion mit Ausbreitung auf die linke hintere Schläfenregion registriert.
Zentrum für Pädiatrische Neurologie und Epilepsie
unter der Leitung von Professor K.Yu. Mukhina beschäftigt sich mit der Diagnose und Behandlung von Angststörungen des Nervensystems in Aetei, spezialisiert auf ätische Formen der Epilepsie.
Hauptrichtungen
Aktivitäten:
Epilepsie bei Kindern und Jugendlichen
Kopfschmerzen
Schlafstörungen bei Kindern
Tiki, Enuresis
Untersuchung von Kindern in den ersten ^ Lebensmonaten.
Untersuchungen in unserem Zentrum:
Diagnose und Behandlung von Erkrankungen des Nervensystems bei Kindern
Vollständige Diagnostik (einschließlich präoperativer) und Behandlung von Epilepsie
Konsultation von Neurologen und Epileptologen
Konsultation eines Kinderarztes (häufig kranke Kinder, Gastroenterologie etc.)
Konsultation eines Psychiaters und Psychologen.
Genetische Beratung mit Tests (einschließlich Karyotypisierung)
Video-EEG-Überwachung (in speziell ausgestatteten Räumen des Zentrums oder bei einem Besuch beim Patienten zu Hause)
Computer (digitale) Elektroenzephalographie
UZDG (Ultraschall-Dopplerographie) der Gefäße von Kopf und Hals
Echoenzephalographie (ECHO EG)
Auf unserer Website können Sie die Zeitschrift "Russian Journal of Child Neurology" über das Internet abonnieren.
Ausführliche Informationen zur Arbeit des Zentrums von 10:00 bis 19:00 Uhr telefonisch:
Tel.: (+7495) 983-09-03; (+7926)290-50-30 Tel./Fax: (+7495) 394-82-52
Adresse: St. Borisovskie Prudy, 13, Geb. 2. Internet: www.epileptologist.ru E-Mail: [E-Mail geschützt](für einen detaillierten Anfahrtsplan siehe Website)
BAND IV AUSGABE 3 2009
fokale hemklonische und sekundär generalisierte Anfälle, die seriell wurden und 48 Stunden anhielten. Frizium wurde Topamax in einer Dosis von 10 mg/Tag mit vorübergehender positiver Wirkung zugesetzt.
Ab dem 8. Lebensjahr wurden Schwierigkeiten bei der Aufnahme von Schulmaterial festgestellt; vermindertes Gedächtnis. Es gab erhöhte Müdigkeit, Erschöpfung, Hemmung der geistigen Aktivität. Der Junge wurde launisch, reizbar, negativ; der Stimmungshintergrund hat abgenommen. Ab dem 9. Lebensjahr nahm diese Symptomatik zu.
Aus der Lebensanamnese ist bekannt, dass das Kind aus einer zweiten normalen Schwangerschaft, einer zweiten Entbindung, einem Geburtsgewicht von 2800 g, einer Länge von 53 cm, einer frühen psychomotorischen und sprachlichen Entwicklung, die voll und ganz dem Alter entsprach, geboren wurde. Frühere Krankheiten: Windpocken im Alter von 6 Jahren, häufige akute Virusinfektionen der Atemwege (bis zu 4 Mal pro Jahr) ab 6 Jahren. Die Vererbung für Epilepsie und andere neurologische Erkrankungen wird nicht belastet.
Zum Zeitpunkt der Untersuchung (11 Jahre alt) war der Zustand des Kindes ernst; reagiert negativ auf die Inspektion. Bewusst, pro-orientiert
Raum und Zeit. Er nimmt äußerst ungern Kontakt auf, weigert sich, Anweisungen zu befolgen. Spontannystagmus nach links, Kopf zur linken Schulter geneigt mit Rechtsdrehung. Die Zunge befindet sich in der Mittellinie, der Pharynxreflex ist reduziert; Dysphagie und Dysarthrie werden festgestellt. Das Sehvermögen ist reduziert.
Es wird eine mäßige diffuse Muskelhypotonie festgestellt. Sehnenreflexe werden gleichmäßig reduziert. Es gab eine leichte Abnahme der Muskelkraft in den rechten Gliedmaßen. Pathologische Fußreflexe wurden nicht festgestellt. Es gibt keine objektiven Daten für die Verletzung der Empfindlichkeit. Lohnt sich im Romberg-Test nicht. Weigert sich zu gehen. Wenn Sie versuchen, ihn auf die Beine zu stellen, weint er, setzt sich auf den Boden. Fehlt bei der Durchführung eines Fingerindex-Tests. Spricht langsam, in einzelnen Worten, widerwillig.
Zusätzliche Untersuchungsmethoden. Video-EEG-Überwachung (2004). Erhebliche Verlangsamung der Hauptaufzeichnungsaktivität im Hintergrund. Während der Studie wurde eine fortgesetzte epileptiforme Aktivität in der linken Okzipitalregion mit Ausbreitung auf die linke hintere Schläfenregion und mit periodischer Bildung eines EEG-Musters aufgezeichnet
geboren 1993 16.12.05
Reis. 2. Patient Ch.A. 11 Jahre. Diagnose: MELAS-Syndrom. Symptomatische fokale Epilepsie.
Video-EEG-Überwachung wurde in der Dynamik nach 1 Jahr (2005) durchgeführt: eine signifikante Verlangsamung der Hintergrundaktivität des Gehirns. Während der Schlafaufzeichnung wird im rechten Fronto-Zentralbereich eine fortgesetzte regionale Verlangsamung aufgezeichnet, in deren Struktur im rechten Fronto-Zentralbereich Peak-Wave-Aktivität festgestellt wird.
ORIGINALARTIKEL
Stupa (Abb. 1). Außerdem wird die fortgesetzte regionale Verzögerung im rechten Fronto-Zentralbereich unter Einbeziehung einzelner scharfer Wellen bestimmt.
Video-EEG-Überwachung in der Dynamik (2005): Signifikante Verlangsamung der Hintergrundaktivität des Gehirns. Die Studie verzeichnete eine anhaltende regionale Verlangsamung in der rechten Fronto-Zentralregion. In der Struktur der regionalen Verzögerung im rechten fronto-zentralen Bereich zeigt sich Peak-Wave-Aktivität (Abb. 2).
MRT des Gehirns. Das erste MRT (6 Jahre) zeigte ein einzelnes hyperintensives Signal im T2-Modus in der linken Hemisphäre des Kleinhirns. MRT-Studie im Laufe der Zeit (10,5 Jahre): Eine signifikante Verschlechterung der primären Läsion wurde mit der Ausbreitung des pathologischen Prozesses weit in die linke und rechte okzipital-parietale Region beider Gehirnhälften (Professor A. A. Alikhanov) festgestellt.
Visuell evozierte Potentiale: Signifikante morphologische und funktionelle Veränderungen im visuell afferenten System auf Höhe des Sehnervs und des kortikalen Teils des visuellen Analysators, stärker ausgeprägt auf der linken Seite.
Augenärztliche Beratung: partielle Atrophie der Sehnerven. Elemente der kortikalen Agnosie.
Elektrokardiogramm: ektopischer Rhythmus mit Beschleunigung bis zu 100 Schlägen pro Minute.
Vertikale Position der elektrischen Achse des Herzens. Veränderungen in Repolarisationsprozessen, die bei Orthostase stärker ausgeprägt sind.
Elektroneuromyographie: zeigte den primären Muskeltyp der Läsion. Die Leitungsgeschwindigkeiten entlang der peripheren Nerven werden nicht reduziert.
Die Untersuchung des Laktatspiegels im Blut: Der Laktatgehalt im Blut beträgt 3,0 mmol / l (die Norm liegt bei bis zu 1,8).
Berücksichtigung von epileptischen Anfällen, die von therapieresistenten Okzipitalregionen der Großhirnrinde ausgehen, schlaganfallähnlichen Episoden, Amauroseperioden, kognitivem Verfall, Vorhandensein von hyperintensiven Signalen im Kleinhirn und hinteren Regionen der Großhirnrinde im MRT , ein Anstieg des Laktatspiegels im Blut, wurde der Patientin eine Diagnose des MELAS-Syndroms nahegelegt. Bei einer genetischen Untersuchung wurde die A3243G-Mutation im heteroplasmatischen Zustand in den Blutzellen gefunden (die Diagnose wurde am Moskauer staatlichen Forschungszentrum der Russischen Akademie der medizinischen Wissenschaften durchgeführt) und die Diagnose wurde verifiziert.
Die Beobachtung in der Nachsorge zeigte ein rasches Fortschreiten von Verletzungen höherer psychischer Funktionen, die Entwicklung einer kortikalen Blindheit, eine vollständige Immobilität des Patienten, gefolgt vom Eintritt des Todes im Alter von 12 Jahren und 10 Monaten. (nach 7 Jahren nach Ausbruch der Krankheit).
Literaturverzeichnis
1. Nikolaeva E.A., Temin P.A. Mitochondriale Erkrankungen, begleitet von einer gestörten neuropsychischen Entwicklung. MELAS-Syndrom // Erbliche Störungen der neuropsychischen Entwicklung von Kindern. Ein Leitfaden für Ärzte, herausgegeben von Temin P.A. Kazantseva L.Z. - Medizin, 2001. - S. 96-107.
2. Nikolaeva E.A., Temin P.A., Nikanorova M.Yu., Klembovsky A.I., Sukhorukov V.S., Dorofeeva M.Yu., Korsunsky A.A. Behandlung eines Kindes mit mitochondrialem Syndrom MELAS (mitochondriale Enzephalopathie, Laktatazidose, Schlaganfall-ähnliche Episoden) // Russian Bulletin of Perinatology and Pediatrics. - 1997. - Nr. 2. - S. 30-34.
3. Smirnova I. N., Kistenev B. A., Krotenkova M. V., Suslina ZA. Schlaganfallartiger Verlauf der mitochondrialen Enzephalomyopathie (MELAS-Syndrom) // Atmosfera. Nervenkrankheiten. - 2006. - Nr. 1. - S. 43-48.
4. Temin PA, Nikanorova M.Yu., Nikolaeva E.A. MELAS-Syndrom (mitochondriale Enzephalomyopathie, Laktatazidose, Schlaganfall-ähnliche Episoden): Hauptmanifestationen, diagnostische Kriterien, Behandlungsmöglichkeiten // Nevrol. Zeitschrift - 1998. - Nr. 2. - S. 43-48.
5. Ajmone-Marsan C., Ralston B. Der epileptische Anfall, seine funktionelle Morphologie und diagnostische Bedeutung. - Springfield (IL): Charles C. Thomas, 1957. - S. 3-231.
6. Aldrich M. S., Vanderzant C. W., Alessi A. G., Abou-Khalil B., Sackellares J. C. Iktale kortikale Blindheit mit dauerhaftem Sehverlust // Epilepsie. - 1989. - V. 30. - S. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. Ein Fall von MELAS mit komplexem Teilstatus epilepticus // Rinsho Shinkeigaku. - 2001. - V. 41(8). - S. 487-90.
BAND IV AUSGABE 3 2009
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptic Phänotypen im Zusammenhang mit mitochondrialen Erkrankungen // Neurologie. - 2001. - V. 56(10). - S. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. Valproinsäure verschlimmert Epilepsie aufgrund von MELAS bei einem Patienten mit einer A3243G-Mutation der mitochondrialen DNA // Metab Brain Dis. - 2007 - V. 22(1). - S. 105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. et al. Molekulare Pathologie von MELAS und MERRF. Die Beziehung zwischen Mutationslast und klinischen Phänotypen // Gehirn. - 1997. - V.120. - S. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Polymorphismus der Epilepsie im Zusammenhang mit der A3243G-Mutation der mitochondrialen DNA (MELAS): Gründe für die verzögerte Diagnose // Rev Neurol (Paris). - 2004. - V. 160 (8-9). - S. 824-829.
12. Dvorkin G., Andermann F., Carpenter S. Klassische Migräne, hartnäckige Epilepsie und multiple Schlaganfälle: ein Syndrom im Zusammenhang mit mitochondrialer Enzephalopathie / In: Andermann F., Lugaresi E., Herausgeber. Migräne und Epilepsie. - Boston: Butterworths, 1987. - S. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Ergebnisse von seriellen Elektroenzephalogrammen bei Patienten mit MELAS // Pediatr Neurol. - 1999. - V. 20(1). - S. 43-48.
14. Goto Y., Nonaka I., Horai S.A. Eine Mutation im tRNA leu(UUR)-Gen, die mit der MELAS-Untergruppe der mitochondrialen Enzephalomyopathien assoziiert ist // Nature. - 1990. - V. 348. - S. 651-653.
15. Hasuo K., Tamura S., Yasumori K., Uchino A., Goda S., Ishimoto S., et al. Computertomographie und Angiographie bei MELAS (mitochondriale Myopathie, Enzephalopathie, Laktatazidose und Schlaganfall-ähnliche Episoden): Bericht von 3 Fällen // Neuroradiologie. - 1987.-V. 29. - S. 393-397.
16. Hirano M., Pavlakis S.G. Mitochondriale Myopathie, Enzephalopathie, Laktatazidose und Schlaganfall-ähnliche Episoden (MELAS): Aktuelle Konzepte // J. clin. Neurol. - 1994. - V. 9. - S. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto, Hirose G. Epileptische Anfälle bei einem Patienten mit mitochondrialer Myopathie, Enzephalopathie, Milchsäure und Schlaganfall-ähnlichen Episoden ( MELAS) // Jpn J Psychiatry Neurol. - 1989. - V. 43(3). - S. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J., et al. Mitochondriale Enzephalomyopathie mit Laktat-Pyruvat-Erhöhung und Hirninfarkt // Neurologie. - 1984. - V. 34. - S. 72-77.
19. Kuzniecky R. Symptomatische Okzipitallappenepilepsie // Epilepsie. - 1998. - V. 39 Suppl 4. - S. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Tiefe und direkte kortikale Aufzeichnung bei Anfallsleiden extratemporalen Ursprungs // Neurologie. - 1976. - V. 26. - S. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Klinische iktale Muster bei Epilepsiepatienten mit okzipitalen elektroenzephalographischen Herden // Neurologie. - 1975. - V. 25. - S. 463-471.
22. Matthews P. M., Tampieri D., Berkovic S. F., Andermann F., Silver K., Chityat D., et al. Die Magnetresonanztomographie zeigt spezifische Auffälligkeiten beim MELAS-Syndrom // Neurologie. - 1991. - V. 41. - S. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. Ein Fall mit MELAS in Verbindung mit Epilepsie partialis continua // Nein zu Hattatsu. - 1991. - V. 23(1). - S. 65-70.
24. Montagna P., Gallassi R., Medori R., Govoni E., Zeviani M., Di Mauro S., et al. MELAS-Syndrom: charakteristische migräneartige und epileptische Merkmale und mütterliche Übertragung // Neurologie. - 1988. - V. 38. - S. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N., et al. Zerebraler Blutfluss bei mitochondrialer Myopathie, Enzephalopathie, Laktatazidose und schlaganfallähnlichen Episoden // Schlaganfall. - 1993. - V. 24. - S. 304-309.
26. Pavlakis S. G., Phillips P. C., Di Mauro S. et al. Mitochondriale Myopathie, Enzephalopathie, Laktatazidose und Schlaganfall-ähnliche Episoden: Ein unverwechselbares klinisches Syndrom // Ein Neurol. - 1984. - V. 16. - S. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. Merkmale des Status epilepticus bei MELAS. Analyse von vier Fällen // Neurologie. - 2006. - V. 21(1). - S. 1-11.
28. Williamson P.D., Spencer S.S. Klinische und EEG-Merkmale komplexer partieller Anfälle extratemporalen Ursprungs // Epilepsie. - 1986. - V. 27 (Beilage 2). - S. 46-63.
29. Williamson P.D., Thadani V.M., Darcey T.M., Spencer D.D., Spencer S.S., Mattson R.H. Okzipitallappenepilepsie: klinische Merkmale, Anfallsausbreitungsmuster und Operationsergebnisse // Ann Neurol. - 1992. - V. 31. - S. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Paradoxe Wirkung von Natriumvalproat, die MELAS-Epilepsie bei einem Patienten mit A3243G-Mutation der mitochondrialen DNA verschlimmert // Central European Journal of Medicine. - 2007. - V. 2(1). - S.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. Vasogenes Ödem bei MELAS: eine serielle Studie mit diffusionsgewichteter MR-Bildgebung // Neurologie. - 1999. - V. 53. - S. 2182-2184.
Das MELAS-Syndrom (MELAS) wurde erstmals 1984 von S. Pavlakis und Kollegen beschrieben. Einige Forscher glauben jedoch, dass das Syndrom bereits früher mit Begriffen wie familiäre Polyodystrophie, Laktatazidämie bezeichnet wurde.
Wesen der Pathologie
1994 beschrieben S. Pavlakis und Mizio Hirano 110 Fälle der Krankheit. MELAS (Mitochondriale Enzephalomyopathie, Laktatazidose und Schlaganfall-ähnliche Episoden) ist eine progressive neurodegenerative Multisystemerkrankung. Es ist polymorph und ist gekennzeichnet durch Enzephalopathie mit Krämpfen und Demenz, Laktatazidose. Die Krankheit wird durch Mutationen in der mitochondrialen DNA (mtDNA) verursacht. Diese Krankheit hat einen anderen Namen - mitochondriale Enzephalomyopathie.
Allgemeine Information
25 bis 44 % der Krankheitsfälle sind erblich und werden über die Mutterlinie übertragen. In anderen Fällen tritt es zum ersten Mal auf. Derzeit sind mehr als 10 Gene bekannt, die mutieren und zur Entstehung dieses Syndroms führen. Dies sind Gene, die die Funktionen der Transfer-RNA kodieren. Das MELAS-Syndrom bezeichnet mitochondriale Erkrankungen (MD) mit einer abnormalen Ansammlung von Mitochondrien, die zu einer Störung des gesamten Energiestoffwechselsystems der Zelle führt.
Krankheiten dieser Gruppe werden nur über die Mutterlinie übertragen. Dabei sind die energieabhängigsten Organe und Gewebe in unterschiedlichen Kombinationen betroffen: Skelettmuskulatur, Herz, Gehirn, Sehkraft, Leber und Nieren.
Die Symptome sind polymorph und können in jedem Alter auftreten. Es umfasst Manifestationen von Diabetes, Hörverlust, Krampfanfällen, Endokrinopathien, Kleinwuchs, Herzerkrankungen, absolute Bewegungsunfähigkeit und psychomotorische Anomalien.
Davor ist die psychomotorische Entwicklung völlig normal. Es wurden keine Patienten mit den gleichen Symptomen identifiziert, da die Mutation viele Gene betrifft: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTTS2, MTND1, 5, 6. Ihre Zahl nimmt weiter zu.
Bei 80 % der Patienten wird das MELAS-Syndrom durch eine Punktsubstitution A3243G im Leucin-tRNA-Gen (UUR) verursacht.
Die Häufigkeit wurde unzuverlässig untersucht. Es gibt nur wenige Daten: In Finnland betrug die A3243G-Mutationsrate beispielsweise 16:100.000 der Bevölkerung; in England - 1 Fall pro 13.000 Menschen.
Pathologische Veränderungen

Ein charakteristisches pathologisches Merkmal des MELAS-Syndroms sind Ragged Red Fibers (RRF), die in Muskelgewebe mit einer speziellen Gomory-Trikolore zu sehen sind. Sie sind das Ergebnis mutierter Gene und das morphologische Substrat von mtDNA-Schäden, die als Ergebnis der Proliferation dieser abnormalen Mitochondrien gebildet werden.
Was ist überhaupt ein Mitochondrium
Mitochondrien sind ein Zweimembran-Organell einer eukaryotischen Zelle (einer Zelle, die einen Zellkern hat), deren Hauptfunktion darin besteht, Energie zu liefern. Das heißt, Mitochondrien sind die Energiebasis der Zellen, ihre Energiestationen.
Die Anzahl der Mitochondrien in Zellen kann im Laufe ihres Lebens von wenigen bis zu Tausenden variieren. Und mehr von ihnen passieren in Zellen, die mit der Energieproduktion verbunden sind.
Die Mitochondrien selbst sind meist rund-länglich und haben eine Größe von 1 bis 10 Mikrometer. Sie können bewegungslos einfrieren oder sich im Zytoplasma der Zelle bewegen. Sie bewegen sich normalerweise dorthin, wo mehr Energie benötigt wird.
Auf der inneren Membran der Mitochondrien befinden sich Auswüchse (Cristae), auf denen sich ganze Enzymsysteme befinden. Grundsätzlich handelt es sich dabei um Eiweißverbindungen. Die Anzahl der Cristae hängt von der Intensität der Syntheseprozesse ab. In den Mitochondrien von Muskelzellen gibt es zum Beispiel immer viele davon.
Mitochondrien haben ein autonomes Proteinsynthesesystem - DNA, RNA und Ribosomen. Einige der notwendigen Proteine werden von den Mitochondrien selbst synthetisiert - 5%, und einige werden aus dem Zytoplasma gewonnen - 95%. Durch eine Vielzahl enzymatischer Reaktionen wird aus organischen Verbindungen Energie gewonnen.
Einige dieser Reaktionen finden unter Beteiligung von Sauerstoff statt, d. h. es findet eine Oxidation statt, und nach anderen wird CO 2 unter Übertragung von Wasserstoffprotonen und Energiefreisetzung freigesetzt. Mit anderen Worten, das Mitochondrium ist ein aktiver Teilnehmer an der Zellatmung.
Diese Reaktionen treten an den Cristae oder in den Mitochondrien selbst auf, was für die Zelle so wichtig ist, dass die Zelle im Falle einer Heilung vollständig gesund ist.
Pathogenese

Beim MELAS-Syndrom ähnelt der Zustand auf den ersten Blick einer Variante nach Schlaganfall. Tatsächlich ist es jedoch untypisch: Es tritt bei jungen Menschen auf, oft provoziert durch Infektionskrankheiten, und kann in Form von bösartigen migräneartigen Kopfschmerzen und Krämpfen auftreten.
Die Angiographie gibt keine vaskulären Pathologien an. Es können normale Gefäße oder vergrößerte Kaliber einiger Arterien, Venen oder Kapillarhyperämie auftreten.
Die MRT zeigt, dass akute Hirnschäden beim MELAS-Syndrom wandern und sogar verschwinden können. Einige Schwerpunkte schwanken. Für einen typischen Schlaganfall ist dies völlig uncharakteristisch.
Beim MELAS-Syndrom liegt eine multifokale Nekrose vor. Dies macht sich hauptsächlich im okzipitalen Teil (posteriore Lokalisation) der Großhirnrinde und der weißen Substanz des Subcortex bemerkbar. Sie können aber auch in anderen Teilen des Gehirns auftreten. Diese Bereiche ähneln einer Nekrose bei einem Herzinfarkt, befinden sich jedoch außerhalb der Becken der zentralen Hirngefäße.
Symptomatische Manifestationen

Normalerweise tritt das MELAS-Syndrom bei Kindern im Alter von 6-10 Jahren auf (es kann mit 3 Jahren und mit 40 Jahren beginnen). Ein früher Beginn der Krankheit ist typischer und betrifft 90 % der Patienten. Bei einem frühen Beginn verläuft die Krankheit schwieriger. Die Patienten sind meist unterdimensioniert, muskulös schwach und absolut nicht an körperliche Anstrengung angepasst.
Jede Anspannung oder körperliche Aktivität führt dazu, dass Sie sich schlechter fühlen. Von den inneren Organen ist das Herz von Muskel- und Leitungsunterernährung betroffen, gefolgt von der Entwicklung einer kardiovaskulären Insuffizienz. Nephropathie, Diabetes, Magen-Darm-Störungen mit Erbrechen treten ebenfalls auf, das Gehör ist reduziert. Gekennzeichnet durch Muskelschmerzen, fehlende Reflexe, Paresen, Krämpfe, IPE, Bewusstlosigkeit. Muskelschwäche (myopathisches Syndrom) und sensorineuraler Hörverlust sind ebenfalls typisch für diese Pathologie.
Endokrinopathie wird nicht nur durch Diabetes mellitus, sondern auch durch Wachstumshormonmangel repräsentiert. Herz- und Nierenerkrankungen sind im Krankheitsverlauf selten.
Anfälle beim MELAS-Syndrom sind sehr variabel. Sie können fokal, generalisiert, tonisch-klonisch und myoklonisch sein. Charakteristisch ist die absolute Unempfindlichkeit der Anfälle gegenüber einer antikonvulsiven Therapie. Oft kommt es vor, dass Ärzte eine Epilepsie diagnostizieren und beispielsweise Valproinsäure verschreiben. Danach verschlechtert sich der Gesundheitszustand stark und die Krämpfe nehmen zu, weil die Mitochondrien unterdrückt werden. Obwohl sich eine Demenz entwickelt, wird sie selten zu einem manifesten Symptom.
Es ist auch charakteristisch für die Krankheit, tritt aber auch bei vielen anderen Pathologien auf und kann daher nicht als Grundlage für die Diagnose dienen. Erst in Kombination mit Migräne, Krämpfen und/oder schlaganfallähnlichen Erscheinungen besteht Verdacht auf das Auftreten des MELAS-Syndroms. Selbst solch ausgedehnte Symptome geben keine korrekte Diagnose. Der Ablauf des Prozesses erfolgt auf unterschiedliche Weise.
Zeichen

Das charakteristische klinische Merkmal des MELAS-Syndroms sind schlaganfallähnliche Episoden (IPE), bei denen plötzlich neurologische Symptome auftreten. IPE ist durch eine Asymmetrie der Läsionen gekennzeichnet. Sie können mehrere sein.
Die Selektivität einer solchen Lokalisierung führt auch zu bestimmten Herdsymptomen:
- Hemianopsie (kortikale Blindheit);
- Hemiparese;
- sensorische Aphasie (Missverständnis von Wörtern);
- Akalkulie (Verletzungen des Kontos);
- Agraphie (Rechtschreibfehler);
- Ataxie (gestörte Koordination willkürlicher Bewegungen);
- Veränderungen im Bewusstsein.
Es ist nicht ungewöhnlich, dass diese Schlaganfall-ähnlichen Symptome alle 1 bis 3 Monate wiederkehren. Die Besonderheit akuter Episoden bei MELAS ist, dass sie eine schnelle Regression haben, aber oft wiederkehren, das heißt, als würden sie spurlos vorübergehen. Darüber hinaus lagern sich bei Patienten mit dieser Erkrankung Verkalkungen in den Basalganglien ab (dies wird im CT festgestellt).
Schlaganfall-ähnliche Episoden entwickeln sich oft im Alter von 5-15 Jahren. Sie werden nie das Ergebnis einer Thromboembolie. Angiopathie bei MELAS ist auf eine Hyperproliferation derselben Mitochondrien zurückzuführen.
IPE in Symptomen manifestiert sich durch wiederkehrende Anfälle von Kopfschmerz, Schwindel, Parese, Lähmung der Gliedmaßen, Hirnnerven. Der Mann ist völlig demoralisiert.
Laktatazidose beim MELAS-Syndrom

Die Hauptursache ist ein Überschuss an Milchsäure im Blut und im Gewebe des Nervensystems. Dadurch wird der Säuregehalt des Blutes in den Arterien stark reduziert. Eine solche Azidose ist ein häufiger Begleiter von Diabetes mellitus, der beim MELAS-Syndrom vorhanden ist.
In einem frühen Stadium sind die Manifestationen unspezifisch. Folgende Symptome werden beobachtet: allgemeine Schwäche, Brustschmerzen, Apathie, Schläfrigkeit. Sehr charakteristisch sind Muskelschmerzen nach körperlicher Anstrengung und intermittierendes schnelles Atmen ohne Geruch.
Im mittleren Stadium reichert sich Milchsäure an und es kommt zum Hyperventilationssyndrom (HVS). Kohlendioxid reichert sich im Blut an. Lautes Atmen beginnt sich zu bilden - Kussmaul. Druckabfall bis zum Kollaps, Oligurie tritt ein. Der Patient wird unruhig, wahnsinnig und verliert dann das Bewusstsein mit der anschließenden Entwicklung eines Komas - dies ist das letzte Stadium. Die Symptome einer Laktatazidose entwickeln sich schnell, nämlich innerhalb weniger Stunden. Dann kommt der Tod.
Diagnostische Maßnahmen

Wie bereits erwähnt, ist die Diagnose des MELAS-Syndroms aufgrund des Polymorphismus der Symptome und der Mutation einer großen Anzahl von Genen schwierig. Gehaltenen:
- allgemeine und biochemische Blutuntersuchungen;
- Muskelbiopsie;
- genetische Studie mit vergleichender Analyse bei erkrankten Angehörigen;
- CT des Gehirns: Infarktbereiche häufiger in den Hemisphären, seltener im Kleinhirn, Basalganglien;
- eine Zunahme des Kalibers der Blutgefäße (Arterien, Venen, Kapillaren);
- DNA-Diagnostik: Suche nach charakteristischen Punktmutationen in mtDNA.
Therapiemethoden
Eine Behandlung des MELAS-Syndroms wurde noch nicht entwickelt und ist derzeit nicht heilbar. Es gibt nur Versuche, den Niederlagesprozess zu verlangsamen. Die Behandlung geht in zwei Richtungen: postsyndromale Therapie (Epilepsie, Diabetes mellitus) und pathogenetische. Allerdings gibt es heute keine wirksame pathogenetische Therapie.
Es gibt symptomatische Behandlungen: Bei Hörverlust werden Hörgeräte aktiv eingesetzt, bei Atemmuskelschwäche wird eine Atemtherapie durchgeführt. Es wurde festgestellt, dass beim MELAS-Syndrom im Blut des Patienten der L-Arginin-Spiegel während der IPE signifikant reduziert ist. Daher wird die Therapie mit Argininpräparaten und Vitaminen durchgeführt. Die positive Wirkung von Coenzym Q oder Idebinon (Noben), Bernsteinsäurepräparaten, Vitamin K 1 und K 3, B 2 , B 3 , E, C wird untersucht; L-Carnitin, Antioxidantien (Mexidol, Mildronat), Korrektoren für Laktatazidose (Dimephosphon) – sie alle verbessern den Energiestoffwechsel der Zelle. Bei der Behandlung von Krampfanfällen werden Valproate und Barbiturate nicht verschrieben, da sie die Mitochondrien unterdrücken.
Um diesem Syndrom vorzubeugen, greifen Sie am besten auf die IVF-Methode zurück. Wenn eine Frau weiß, dass sie eine Manifestation dieser Krankheit in ihrer Familie hat, wird das Zytoplasma für die Befruchtung einer gesunden Frau entnommen. Die Methode befindet sich noch im Studienstadium, sie ist keine Masse.
Einige Eigenschaften
Die Diagnose von mitochondrialen Erkrankungen erfordert einen sehr sorgfältigen Therapieansatz. Mittel zur Stoffwechselwirkung müssen unbedingt darin enthalten sein. Sie stabilisieren die Prozesse der Gewebeatmung, oxidative Phosphorylierung in Zellen. Nur die systematische Umsetzung einer solchen Behandlung kann dazu beitragen, den Zustand der Patienten zu erhalten und Schlaganfällen vorzubeugen.
Vorhersage
Die Prognose ist aufgrund des Fehlens einer wirksamen Behandlung ungünstig. Die Lebenserwartung ab dem ersten Auftreten der Symptome beträgt in der Regel nicht mehr als fünf Jahre. Die mangelnde Kenntnis der Krankheitsursachen führt dazu, dass das optimale Behandlungsschema noch nicht gefunden wurde. All dies macht die Heilungschancen minimal.
Das MELAS-Syndrom ist eine mitochondriale Erkrankung, die durch Muskel- und ZNS-Beteiligung gekennzeichnet ist.
MELAS (eng. Mitochondriale Enzephalomyopathie, Laktatazidose und Schlaganfall-ähnliche Episoden - „Mitochondriale Enzephalomyopathie, Laktatazidose, Schlaganfall-ähnliche Episoden“) ist eine fortschreitende neurodegenerative Erkrankung, die durch die im Titel aufgeführten Manifestationen gekennzeichnet ist und von polymorphen Symptomen begleitet wird - Schlaganfall, Diabetes, Krampfanfälle, verringerter Hörverlust, Herzerkrankungen, Kleinwuchs, Endokrinopathien, Belastungsintoleranz und neuropsychiatrische Störungen.
Geschichte.
Das MELAS-Syndrom wurde erstmals 1984 von Pavlakis und Kollegen beschrieben; Zehn Jahre später veröffentlichten Pavlakis und Mizio Hirano eine Übersicht über 110 Fälle.
Vererbungsart:
mütterlich
Epidemiologie:
Die genaue Häufigkeit der Erkrankung ist nicht bekannt. In der Literatur gibt es nur wenige Angaben zur Häufigkeit der Erkrankung. In Nordfinnland beträgt die A3243G-Mutationsrate 16,3:100.000.
Pathogenese:
Mutationen der mitochondralen DNA, die die Atmungskette der Mitochondrien steuern, gehen einher mit einer Störung der Prozesse der oxidativen Phosphorylierung, der wichtigsten Energiequelle für Stoffwechselvorgänge in der Zelle.
Klinische Manifestationen
Im Alter von 40 Jahren werden Patienten mit MELAS mit einer vorübergehenden ischämischen Attacke sowie mit Epilepsie, wiederholtem Erbrechen, Kopfschmerzen und Muskelschwäche in eine Klinik eingewiesen. Bei diesen Patienten wird häufig eine Demenz klinisch diagnostiziert.
Das junge Alter und das Fehlen schlaganfallspezifischer Risikofaktoren machen MELAS nachdenklicher.
Labordaten
Laktatazidose - erhöhte Laktat- und Pyruvatspiegel.
Visualisierungsdaten
Veränderungen im Gehirn ähneln Veränderungen bei einem Schlaganfall.
Unterschiede zu einem Schlaganfall
1) Die betroffenen Bereiche stimmen nicht mit den Grenzen der arteriellen Gefäßbecken überein.
2) bei wiederholten Angriffen werden die Herde in einer anderen Lokalisation visualisiert.
+ klinische Daten (junges Alter, keine Risikofaktoren für Schlaganfall).
CT
Mehrere hypodense Bereiche, die nicht mit dem Gefäßbett übereinstimmen.
Verkalkung der Basalganglien (am häufigsten bei älteren Patienten).
Atrophie tritt vor dem Hintergrund von Regression und klinischer Besserung auf.
MRT
Akuter Infarkt
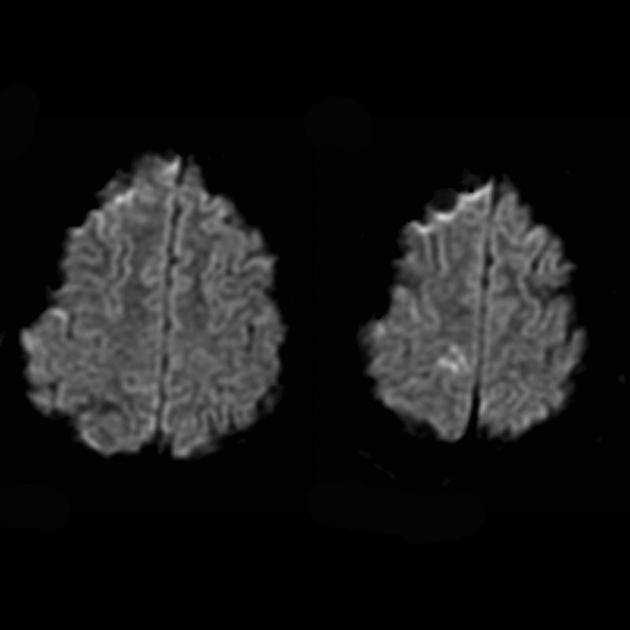
Zur Differenzierung beim Schlaganfall werden ADC und DWI verwendet (Diffusionseinschränkung beim Schlaganfall (zytotoxisches Ödem), bei MELAS ist die Diffusion leicht eingeschränkt oder unverändert (vasogenes Ödem).
Beteiligung am pathologischen Prozess der subkortikalen weißen Substanz des Gehirns.
Verschlechterung der Visualisierung der Klarheit der Konturen der Windungen und eine Zunahme des Signals von ihnen auf T2-gewichteten Bildern.
Chronischer Infarkt
Änderungen können symmetrisch oder asymmetrisch sein.
Fokale Atrophie tritt vor dem Hintergrund von Regression und klinischer Besserung auf.
Am häufigsten sind der Parietal-, Okzipital- und Temporallappen des Gehirns betroffen.
MR-Spektroskopie
Erhöhte Laktatwerte.




Die Materialien richten sich an Neurologen, Therapeuten und Allgemeinmediziner.
Sergey Likhachev, Leiter, MD. Wissenschaften, Professor;
Inessa Pleshko, Leitende Forscherin, Ph.D. Wissenschaften, Neurologische Abteilung des Republikanischen Wissenschaftlichen und Praktischen Zentrums für Neurologie und Neurochirurgie.
Die zerebrale autosomal-dominante Arteriopathie mit subkortikalen Infarkten und Leukenzephalopathie (CADASIL) ist eine fortschreitende autosomal-dominante Erkrankung, deren klinische Manifestationen rezidivierende subkortikale ischämische Schlaganfälle, Migräne, subkortikale Demenz und affektive Störungen umfassen. Aktuelle Prävalenz - 1 Fall
pro 100.000 Einwohner.
Das Republican Scientific and Practical Center for Neurology and Neurosurgery behandelt 7 Patienten (darunter 4 Frauen) mit CADASIL; Alter - von 32 bis 68 Jahren. Sie wurden mit neurologischen, molekulargenetischen Methoden untersucht. Es gab charakteristische Symptome; in der Geschichte - Migräne, wiederkehrende lakunare Schlaganfälle und affektive Störungen. Die MRT des Gehirns zeigte subkortikale Infarkte und eine für CADASIL charakteristische Leukenzephalopathie.
Als Ergebnis der molekulargenetischen Diagnostik wiesen 2 Personen eine heterozygote Mutation im Notch3-Gen auf dem 19. Chromosom auf, die CADASIL verursacht. Notch-Gene codieren Transmembranrezeptoren, die an der Zellontogenese beteiligt sind. Bei CADASIL werden in den meisten Fällen Missense-Mutationen festgestellt, durch die sich die Struktur des Transmembranproteins verändert und seine Funktionen beeinträchtigt werden.
Die Pathogenese von CADASIL ist nicht vollständig geklärt. Es wird angenommen, dass der Hauptfaktor eine Arteriopathie mit fortschreitender Okklusion kleiner perforierender Gefäße der weißen Substanz des Gehirns ist (was zu einer chronischen Hypoperfusion führt). Gleichzeitig werden charakteristische körnige osmiophile Einschlüsse gefunden, die eine Proliferation von Basalmembrankomponenten, eine Verdickung der Mittelmembran und eine mechanische Kompression kleiner Arterien verursachen. Dadurch wird die Blut-Hirn-Schranke geschädigt - es entsteht ein Ödem.
Ein zusätzlicher pathologischer Faktor ist die Aktivierung von Astrozyten in der Nähe der Gefäßwand. Sie setzen Endothelium-1 frei, was zu Vasokonstriktion und Beeinträchtigung des Blutflusses führt.
Die Zusammensetzung körniger osmiophiler Einschlüsse ist unbekannt. Es wird angenommen, dass das Notch3-Protein einer ihrer Bestandteile ist. In Hautbiopsien von Patienten mit einer Notch3-Mutation lassen sich bereits vor dem 20. Lebensjahr osmiophile Granula und eine Degeneration glatter Muskelzellen nachweisen.
Klinische Diagnostik von CADASIL:
- belastete Familiengeschichte;
- die Entwicklung der ersten Krankheitssymptome vor dem 50. Lebensjahr;
- das Vorhandensein von zwei der folgenden Symptome - Migräne, wiederkehrende Schlaganfälle, Stimmungsstörungen, subkortikale Demenz.
Vaskuläre Risikofaktoren, die ätiologisch mit neurologischen Symptomen assoziiert sind, sollten ausgeschlossen werden. Die MRT zeigt Schäden an der weißen Substanz der Gehirnhälften und das Fehlen kortikaler Infarkte.
Eine sichere Diagnose von „CADASIL“ wird durch ein positives Ergebnis der molekulargenetischen Diagnostik oder den Nachweis einer Arteriopathie mit charakteristischen granulären osmiophilen Einschlüssen in Haut- oder Muskelbiopsien bestätigt.
Die häufigsten Symptome von CADASIL sind transitorische ischämische Attacken und ischämische Schlaganfälle, die bei fast 85 % der Patienten beobachtet werden.
Sie sind durch einen rezidivierenden Verlauf gekennzeichnet, der sich durch klassische Syndrome des lakunären Schlaganfalls und eine vollständige klinische Remission nach einigen Tagen oder Wochen manifestiert.
Am zweithäufigsten sind kognitive Beeinträchtigungen (bei 60 % der Patienten festgestellt). Kann im Alter von 35 Jahren beginnen, manchmal sogar vor ischämischen Episoden. Etwa 75 % der CADASIL-Patienten entwickeln eine Demenz. Das erste Symptom ist normalerweise eine Migräne; tritt häufig vor dem 20. Lebensjahr auf und geht normalerweise einem Schlaganfall voraus.
Daten zur Beteiligung des Herzens am pathologischen Prozess bei CADASIL sind widersprüchlich. L. Oberstein et al. (2003) fanden heraus, dass 25 % der mit CADASIL diagnostizierten Patienten in der Vorgeschichte einen akuten Myokardinfarkt oder eine Q-Wellen-Pathologie im Elektrokardiogramm hatten. In einer anderen Studie untersuchten Cumurciuc et al. (2006) fanden bei 23 Personen mit einer Notch3-Mutation keine positive Herzanamnese.
Klinische Manifestationen von CADASIL und zerebraler Mikroangiopathie unterschiedlicher Ätiologie sind ähnlich – eine Differenzialdiagnose ist erforderlich.
Um CADASIL bei Patienten und ihren Familien rechtzeitig zu bestimmen, muss auf molekulargenetische Methoden und/oder histologische Untersuchungen zurückgegriffen werden.
MELAS-Syndrom
Die mitochondriale Enzephalomyopathie mit Laktatazidose und schlaganfallähnlichen Episoden (MELAS) ist eine seltene Erbkrankheit, die durch eine Pathologie des mitochondrialen Genoms, eine Beeinträchtigung des Energiestoffwechsels und der Funktion der energieabhängigsten Organe und Gewebe (ZNS, Herz- und Skelettmuskulatur, Augen, Nieren, Leber, Knochenmark, endokrines System). Die große Variabilität der klinischen Manifestationen des MELAS-Syndroms und das seltene Auftreten prädestinieren diagnostische Schwierigkeiten für den Arzt.
Im Republikanischen Wissenschafts- und Praxiszentrum für Neurologie und Neurochirurgie werden 3 Patienten (eine 46-jährige Frau und ihre Söhne im Alter von 24 und 23 Jahren) mit einem diagnostizierten MELAS-Syndrom beobachtet. Sie wurden klinisch und neurologisch untersucht, molekulargenetische Diagnostik, MRT des Gehirns.
Alle sind kurz; in der Geschichte - Symptome der mitochondrialen Pathologie: sensorineuraler Hörverlust, migräneartige Kopfschmerzen, schlechte Belastungstoleranz. Das Debüt der Krankheit sind generalisierte Krampfanfälle. Bei 2 Patienten traten die ersten Symptome vor dem 20. Lebensjahr auf; es gab epileptische Anfälle nacheinander, Episoden von Sehstörungen mit dem Vorhandensein von Herden in der Neurobildgebung im okzipitalen und temporalen Bereich, einen Anstieg des Laktatspiegels im Blut und in der Zerebrospinalflüssigkeit. 1 Person hatte eine mäßige Abnahme der kognitiven Funktionen; laut Ultraschall des Herzens - hypertrophe Kardiomyopathie; Diabetes mellitus.
Eine molekulargenetische Studie ergab für MELAS typische Multisystemläsionen, eine große Variabilität und unterschiedliche Grade klinischer Manifestationen, die der Anzahl von A3243G-Mutantenkopien im tRNA-Leu(UUR)-Gen entsprechen.
MELAS ist durch eine mütterliche Vererbung gekennzeichnet, das Vorhandensein sporadischer Fälle, wenn eine De-novo-Mutation auftritt; Akkumulation von mitochondrialer DNA (Heteroplasmie) in Zellen – sowohl normalen als auch mutierten Typen – und zufällige Verteilung während der Teilung zwischen Tochterzellen (mitotische Segregation). Auf genetischer Ebene ist die Ursache des MELAS-Syndroms das heteroplasmatische Rearrangement 3243A>G im tRNALeu(UUR)-Gen (80 % der Fälle werden erkannt).
Die Pathogenese der Krankheit wurde noch nicht untersucht. Es gibt 2 Haupttheorien - "mitochondriale Angiopathie" und "mitochondriale Zytopathie". Es ist bekannt, dass die schlaganfallartige Läsion nicht den Gefäßzonen entspricht und sich aufgrund eines begleitenden vasogenen Ödems, das durch eine anhaltende epileptische Aktivität verursacht wird, auf die umgebenden Bereiche ausdehnt. Wie vermutet, sind schlaganfallähnliche Episoden auf eine neuronale Übererregbarkeit in einem begrenzten Bereich des Gehirns zurückzuführen. Es entsteht durch mitochondriale Dysfunktion in kapillaren Endothelzellen oder in Neuronen oder in Astrozyten; depolarisiert benachbarte Neuronen, was zur Ausbreitung epileptischer Aktivität führt.
Darüber hinaus haben Patienten mit MELAS in den Intervallen zwischen Schlaganfall-ähnlichen Episoden laut Single-Photon-Emissions-Computertomographie (SPECT) eine Minderdurchblutung des hinteren cingulären Kortex, was auf eine Störung der zerebralen Hämodynamik hinweist.
Eine Verletzung der oxidativen Phosphorylierung, ein Bruch der mitochondrialen Atmungskette tragen zur Dominanz des katabolen Stoffwechsels und zu einem Wechsel vom Krebszyklus zur anaeroben Glykose mit Laktatakkumulation bei. Ein hoher Spiegel des letzteren im ZNS korreliert normalerweise mit Perioden neurologischer Symptome.
Die wichtigsten klinischen Anzeichen von MELAS sind schlaganfallähnliche Episoden, Laktatazidose und das Vorhandensein von "zerrissenen roten Fasern" in Muskelbiopsieproben. Zusätzliche Manifestationen können Demenz, Psychose, epileptische Anfälle, migräneartige Kopfschmerzen, Ataxie, Myopathie, Verkalkung der Basalganglien in der Neuroimaging, optische Atrophie, Retinopathie, Taubheit, Diabetes, intestinale Pseudoobstruktion, Kardiomyopathie sein.
Das frühe Alter des MELAS-Debüts liegt zwischen 5 und 20 Jahren, es gibt jedoch Beobachtungen eines späten Beginns - in der 5. bis 6. Lebensdekade. Es gibt Fälle, in denen das Syndrom nach Herzerkrankungen begann.
Die multisystemische Natur der Läsionen bei MELAS erschwert die klinische Diagnose.
Die erbliche Natur der Krankheit zwingt zur Durchführung molekulargenetischer Untersuchungen, um eine genaue Diagnose stellen zu können.
und andere Patienten identifizieren - unter den Angehörigen des Patienten.
Die Materialien richten sich an Neurologen, Therapeuten und Allgemeinmediziner.
| Mitochondriale Myopathie, Enzephalomyopathie, Laktatazidose und Schlaganfall-ähnliche Episoden | |
|---|---|
| Verkalkung der Basalganglien, Kleinhirnatrophie, erhöhtes Laktat; CT-Bild einer Person, bei der MELAS diagnostiziert wurde | |
| Spezialität | Neurologie |
Genetik
Muskelbiopsie einer Person, bei der MELAS diagnostiziert wurde, die aber keine bekannte Mutation trägt. (a) Gomoris modifizierter dreifarbiger Farbstoff zeigt einige ausgefranste rote Fasern (Pfeile). (b) Cytochrom-c-Oxidase-Färbung, die Typ-1-, leicht gefärbte und Typ-II-Fasern, dunkle Fasern und einige Fasern mit abnormalen mitochondrialen Ansammlungen zeigt (Pfeile). Beachten Sie, dass Cytochrom-C-Oxidase-negative Fasern normalerweise bei mitochondrialer Enzephalopathie, Laktatazidose und Schlaganfall-ähnlichen Episoden (MELAS) zu sehen sind. (c) Succinat-Dehydrogenase-Färbung zeigt mehrere ausgefranste blaue Fasern und intensive Färbung in Blutgefäß-Mitochondrien (Pfeil). (d) Die Elektronenmikroskopie zeigt eine abnormale Ansammlung von Mitochondrien mit parakristallinen Einschlüssen (Pfeile), osmiophilen Einschlüssen (große Pfeile) und mitochondrialen Vakuolen (kleine Pfeile).
MELAS wird durch Mutationen in Genen in der mitochondrialen DNA verursacht.
NADH-Dehydrogenasen
Mutationen ein MT-TL1 verursachen über 80 Prozent aller MELAS-Fälle. Sie reduzieren die Fähigkeit der Mitochondrien, Proteine herzustellen, Sauerstoff zu verbrauchen und Energie zu produzieren. Forscher haben nicht festgestellt, wie Veränderungen in der mitochondrialen DNA zu den spezifischen Anzeichen und Symptomen von MELAS führen. Sie untersuchen weiterhin die Auswirkungen von mitochondrialen Genmutationen in verschiedenen Geweben, insbesondere im Gehirn.
Nachlass
Dieser Zustand wird in einem mitochondrialen Muster vererbt, das auch als mütterliche Vererbung und Heteroplasmie bekannt ist. Dieses Vererbungsmuster bezieht sich auf Gene, die in der mitochondrialen DNA enthalten sind. Da Eier, aber keine Spermien Mitochondrien zum sich entwickelnden Embryo beitragen, durchlaufen nur Frauen für ihre Babys mitochondriale Bedingungen. Mitochondriale Störungen können in jeder Generation einer Familie auftreten und sowohl Männer als auch Frauen betreffen, aber Väter geben die mitochondrialen Merkmale nicht an ihre Kinder weiter. In den meisten Fällen erben Menschen mit MELAS das veränderte mitochondriale Gen von ihrer Mutter. Weniger häufig resultiert die Störung aus einer neuen Mutation im mitochondrialen Gen und tritt bei Menschen ohne eine familiäre Vorgeschichte von MELAS auf.
Diagnose
Behandlung / Prognose
Patienten werden danach behandelt, welche Bereiche des Körpers zu einem bestimmten Zeitpunkt betroffen sind.