Jak długo żyją dzieci z zespołem Melas? Rzadkie choroby. Objawy zespołu MELAS
Słowa kluczowe
ZESPÓŁ MELAS / ZESPÓŁ MELAS / PADACZKA / PADACZKA / KLINIKAadnotacja artykuł naukowy o medycynie klinicznej, autor pracy naukowej - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
Zespół MELAS jest genetycznie uwarunkowaną chorobą z grupy chorób mitochondrialnych, określanych jako encefalomiopatia mitochondrialna z kwasicą mleczanową i epizodami udaropodobnymi (encefalomiopatia mitochondrialna, kwasica mleczanowa z epizodami udaropodobnymi). W proces patologiczny zaangażowane są wszystkie narządy i tkanki, ale w większym stopniu cierpią układ mięśniowy i nerwowy. Choroba najczęściej rozwija się w wieku od 6 do 10 lat. Przebieg choroby postępuje. W większości przypadków choroba objawia się napadami padaczkowymi, nawracającymi bólami głowy, wymiotami i anoreksją. Padaczka jest ważnym objawem klinicznym zespołu MELAS. Napady padaczkowe są pierwszym rozpoznawalnym objawem encefalopatii mitochondrialnych (ME) w 53% przypadków. W MELAS najczęściej występuje padaczka potyliczna. Wraz z postępem choroby odnotowuje się oporność padaczki na terapię, często z przebiegiem stanu. Opisano przypadki transformacji w padaczkę Kozhevnikova. Przedstawiamy historię przypadku pacjenta z rozpoznaniem zespołu MELAS zweryfikowanym za życia.
Powiązane tematy artykuły naukowe z medycyny klinicznej, autor pracy naukowej - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
-
Encefalopatia mitochondrialna z epizodami udaropodobnymi i kwasicą mleczanową (zespół Melas): kryteria diagnostyczne, cechy napadów padaczkowych i sposoby leczenia na przykładzie przypadku klinicznego
2017 / Yamin M.A., Chernikova IV, Araslanova L.V., Shevkun P.A. -
Udary w chorobach mitochondrialnych
2012 / Pizova N.V. -
Padaczka u dzieci z chorobami mitochondrialnymi: cechy diagnozy i leczenia
2012 / Zavadenko N. N., Kholin A. A. -
Zaburzenia neurologiczne w encefalomiopatii mitochondrialnej - kwasica mleczanowa z epizodami udaropodobnymi (zespół MELAS)
2012 / Kharlamov Dmitrij Aleksiejewicz, Krapiwkin Aleksiej Igorewicz, Suchorukow Władimir Siergiejewicz, Kuftina Ludmiła Andriejewna, Groznowa Olga Siergiejewna -
Zespół Melasa jako niezwykła przyczyna niedoczynności przytarczyc: przypadek kliniczny
2018 / Umyarova Dilyara Shamilevna, Grebennikova Tatiana Alekseevna, Zenkova Tatiana Stanislavovna, Sorkina Ekaterina Leonidovna, Zhanna Belaya -
Epizody udaropodobne w encefalomiopatii mitochondrialnej z kwasicą mleczanową
2010 / Kałasznikowa Ludmiła Andrejewna, Dobrynina L. A., Sakharova A. V., Chaikovskaya R. P., Mir-kasimov M. F., Konovalov R. N., Shabalina A. A., Kostyreva M. V., Gnezditsky V. V., Protsky S. V. -
Cytopatie mitochondrialne: melas i zespoły MIDD. Jedna wada genetyczna, różne fenotypy kliniczne
2017 / Muranova A.V., Strokov I.A. -
Łagodna padaczka potyliczna wieku dziecięcego o wczesnym początku (zespół Panayotopoulosa). Opis przypadku klinicznego
2015 / Matyuk Yu.V., Kotov A.S., Borisova M.N., Panteleeva M.V., Shatalin A.V. -
Polimorfizm objawów klinicznych postępującej encefalomiopatii mitochondrialnej związanej z mutacją genu POLG1
2016 / Yablonskaya M.I., Nikolaeva E.A., Shatalov P.A., Kharabadze M.N. -
Wartość diagnostyczna badania aktywności cytochemicznej enzymów w dziedzicznych chorobach mitochondrialnych
2017 / Kazantseva I.A., Kotov S.V., Borodataya E.V., Sidorova O.P., Kotov A.S.
PADACZKA W ZESPOLE MELAS
Zespół MELAS to genetycznie uwarunkowana choroba z grupy mitochondrialnej, definiowana jako encefalomiopatia mitochondrialna, kwasica mleczanowa z epizodami udaropodobnymi. Proces patologiczny obejmuje wszystkie narządy i tkanki, ale w większości jest niekorzystny dla układu mięśniowego i nerwowego. Choroba występuje najczęściej u dzieci w wieku od 6 do 10 lat. Przebieg kliniczny jest postępujący. W większości przypadków choroba objawia się napadami padaczkowymi, nawracającymi bólami głowy, wymiotami, anoreksją. Ważnym obrazem klinicznym zespołu MELAS jest padaczka. Napady padaczkowe są początkowym objawem encefalopatii mitochondrialnych (ME) w 53% przypadków. W zespole MELAS najczęściej występuje padaczka potyliczna. W miarę postępu choroby obserwuje się oporność padaczki na leczenie, często z występowaniem stanu padaczkowego. Opisano kilka przypadków przejścia w padaczkę Kozhevnikova oraz przedstawiono historię pacjenta ze zweryfikowaną za życia diagnozą zespołu MELAS.
Tekst pracy naukowej na temat „Padaczka w zespole melasa”
TOM IV WYDANIE 3 2009
PADACZKA Z ZESPOŁEM MELAS
K.Yu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, CB. Michajłowa2, Wirginia. Czadajew1, AA. Alikhanov 1-2, B.N. Ryżkow1, A.S. Petrukhin1
PADACZKA W ZESPOLE MELAS
KYu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, S.V. Michajłowa2, UA. Czadajew1, AA. Alikhanov 1-2, B.N. Ryzkov1 AS. Petrukhin1
1 - Katedra Neurologii i Neurochirurgii, Wydział Pediatrii, Państwowa Instytucja Edukacyjna Wyższego Szkolnictwa Zawodowego, Rosyjski Państwowy Uniwersytet Medyczny w Roszdrav
2 - Rosyjski Dziecięcy Szpital Kliniczny
Zespół MELAS jest genetycznie uwarunkowaną chorobą z grupy chorób mitochondrialnych, określanych jako encefalomiopatia mitochondrialna z kwasicą mleczanową i epizodami udaropodobnymi (encefalomiopatia mitochondrialna, kwas mlekowy z epizodami udaropodobnymi). W proces patologiczny zaangażowane są wszystkie narządy i tkanki, ale w większym stopniu cierpią układ mięśniowy i nerwowy. Choroba najczęściej rozwija się w wieku od 6 do 10 lat. Przebieg choroby postępuje. W większości przypadków choroba objawia się napadami padaczkowymi, nawracającymi bólami głowy, wymiotami i anoreksją. Padaczka jest ważnym objawem klinicznym zespołu MELA. Napady padaczkowe są pierwszym rozpoznawalnym objawem encefalopatii mitochondrialnych (ME) w 53% przypadków. W MELAS najczęściej występuje padaczka potyliczna. Wraz z postępem choroby odnotowuje się oporność padaczki na terapię, często z przebiegiem stanu. Opisano przypadki transformacji w padaczkę Kozhevnikova. Przedstawiamy historię przypadku pacjenta z rozpoznaniem zespołu MELAS zweryfikowanym za życia.
Słowa kluczowe: zespół MELAS, padaczka, klinika, diagnostyka, leczenie.
Zespół MELAS to genetycznie uwarunkowana choroba z grupy mitochondrialnej, definiowana jako encefalomiopatia mitochondrialna, kwasica mleczanowa z epizodami udaropodobnymi. Proces patologiczny obejmuje wszystkie narządy i tkanki, ale w większości jest niekorzystny dla układu mięśniowego i nerwowego. Choroba występuje najczęściej u dzieci w wieku od 6 do 10 lat. Przebieg kliniczny jest postępujący. W większości przypadków choroba objawia się napadami padaczkowymi, nawracającymi bólami głowy, wymiotami, anoreksją. Ważnym obrazem klinicznym zespołu MELAS jest padaczka. Napady padaczkowe są początkowym objawem encefalopatii mitochondrialnych (ME) w 53% przypadków. W zespole MELAS najczęściej występuje padaczka potyliczna. W miarę postępu choroby obserwuje się oporność padaczki na leczenie, często z występowaniem stanu padaczkowego. Opisano kilka przypadków przejścia w padaczkę Kozhevnikova oraz przedstawiono historię pacjenta ze zweryfikowaną za życia diagnozą zespołu MELAS.
Słowa kluczowe: zespół MELAS, padaczka, obraz kliniczny, diagnostyka, leczenie.
Zespół MELAS jest genetycznie uwarunkowaną chorobą z grupy chorób mitochondrialnych, określanych jako encefalomiopatia mitochondrialna z kwasicą mleczanową i epizodami udaropodobnymi (encefalomiopatia mitochondrialna, kwasica mleczanowa z epizodami udaropodobnymi).
Zespół MELAS został po raz pierwszy zidentyfikowany jako niezależna forma nozologiczna przez S. Pavlakisa i in. w 1984 roku. Jednak wielu autorów sugeruje, że choroba została wcześniej opisana pod nazwą „rodzinna poliodystrofia, miopatia mitochondrialna, kwasica mleczanowa”.
Częstość występowania w populacji nie została ustalona. Do 2000 roku opublikowano ponad 120 obserwacji zespołu MELAS, w tym w prasie krajowej.
Zespół MELAS w 25% przypadków jest dziedziczony po matce z wysokim ryzykiem, ale u 56-75% pacjentek wywiad rodzinny nie jest obciążony. Choroba jest związana z mutacjami w mitochondrialnych genach DNA kodujących podjednostki kompleksów łańcucha oddechowego oraz geny transportowego RNA (MT-ND1, MT-ND5, MT-TH, MT-TL1 i MT-TV). W 80-90% przypadków zespołu MELAS choroba opiera się na mutacji punktowej w genie MT-TL1 kodującym RNA transferu leucyny. Dzięki tej mutacji nukleotyd adeninowy zostaje zastąpiony guaniną w pozycji 3243 (A3243G), co zakłóca syntezę wszystkich białek w mitochondriach.
W proces patologiczny zaangażowane są wszystkie narządy i tkanki, ale w większym stopniu cierpią układ mięśniowy i nerwowy.
Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova C.V., Chadaev V.A., Alikhanov A.A., Ryżkow BN., Petrukhin A.S.
Padaczka w zespole MELAS Rus. żur. det. Neur.: t. IV, nr. 3, 2009.
ARTYKUŁY ORYGINALNE
tematy jako najbardziej zmienne. Nasilenie objawów klinicznych zależy od efektu progowego (wiek, zapotrzebowanie energetyczne tkanek), kontroli genów jądrowych nad syntezą łańcucha oddechowego, heteroplazmii (różna zawartość zmutowanych cząsteczek mtDNA w tkankach). Wykazano, że u pacjentów z zespołem MELAS zawartość zmutowanego mtDNA w różnych tkankach wynosi 93-96%. U członków rodziny probantów zmutowany mtDNA jest również wykrywany w tkankach, ale jego zawartość jest znacznie niższa: 62-89% w wymazanej postaci choroby, od 28 do 89% przy braku objawów klinicznych zespołu.
Choroba rozwija się najczęściej w wieku od 6 do 10 lat, ale zdarzają się przypadki debiutu wcześniejszego (do dwóch lat) lub późniejszego - od 21 do 40 lat. Przed zachorowaniem 90-100% pacjentów rozwija się normalnie. Przebieg choroby jest postępujący, bardziej złośliwy z wczesnym początkiem.
W większości przypadków choroba objawia się napadami padaczkowymi, nawracającymi bólami głowy, wymiotami i anoreksją. Należy również zwrócić uwagę na nietolerancję aktywności fizycznej w postaci pogorszenia stanu zdrowia i pojawienia się osłabienia mięśni. Zespół objawów miopatycznych objawia się nietolerancją wysiłku, osłabieniem mięśni, zmęczeniem, a czasem hipotrofią mięśni.
W miarę postępu choroby zwykle rozwija się demencja. Rzadziej występują takie objawy, jak ataksja móżdżkowa, głuchota neurosensoryczna i polineuropatia obwodowa.
Charakterystyczne są epizody udarowe, które mogą objawiać się nawracającymi atakami bólu głowy, zawrotami głowy, rozwojem ogniskowych objawów neurologicznych (niedowład, hemianopsja) i śpiączką. Te ostre epizody są często wywoływane przez gorączkę lub współistniejące infekcje. Objawy te mogą mieć dość szybką regresję (od kilku godzin do kilku tygodni), a także tendencję do nawrotów.
Padaczka jest ważnym objawem klinicznym, często występującym we wczesnych stadiach MELAS. to
często najbardziej oczywistym objawem neurologicznym, zwłaszcza w atypowej encefalopatii mitochondrialnej (ME). Napady padaczkowe są pierwszym rozpoznawalnym objawem encefalopatii mitochondrialnych (ME) w 53% przypadków.
W MELAS najczęściej występuje padaczka potyliczna (SE). Charakteryzuje się napadami ogniskowymi wywodzącymi się z płatów potylicznych. Napady często wiążą się z przejściowymi lub uporczywymi objawami neurologicznymi, takimi jak utrata pola widzenia.
Napady padaczkowe pochodzące z kory potylicznej dzieli się według ich przejawów na odczucia subiektywne (aura) i objawy wykrywalne klinicznie, z reguły z komponentem motorycznym. Aury padaczkowe emanujące z płata potylicznego obejmują proste i złożone halucynacje wzrokowe, amaurozę. Najbardziej typowymi napadami charakterystycznymi dla SE są proste halucynacje wzrokowe, które mogą objawiać się objawami pozytywnymi (błyski, plamki światła) i negatywnymi (mroczka, hemianopsja). Najczęściej halucynacje wzrokowe są opisywane jako plamka lub plamki światła, stałe lub migające. Z reguły plama jest biała z zielonkawym odcieniem. Ponadto halucynacje mogą być wielokolorowe lub monochromatyczne. Halucynacje zwykle pojawiają się w polach widzenia przeciwnych do ogniska wzbudzenia w korze potylicznej, a następnie rozprzestrzeniają się. Należy jednak zauważyć, że w skargach pacjentów aura wzrokowa nie jest często wykrywana.
Złożone halucynacje wzrokowe obserwuje się, gdy pobudzenie padaczkowe rozprzestrzenia się na regiony potyliczno-skroniowe lub potyliczno-ciemieniowe. Złożone halucynacje wzrokowe mogą pojawiać się w postaci ludzi, obiektów zwierzęcych lub scen, być znajome lub nieznane, przyjemne lub przerażające, przerażające, proste lub groteskowe, mogą być statyczne lub poruszać się w płaszczyźnie poziomej i znikać. Z reguły są one końcowym objawem przed rozwojem ataku motorycznego; może być pierwszym objawem napadu, ale częściej pojawia się po nim
TOM IV WYDANIE 3 2009
podstawowe halucynacje.
Ictal ama vrosis to szczególny, niezwykle trudny do zdiagnozowania rodzaj napadów padaczkowych pochodzących z kory potylicznej. Według wielu autorów jest to ten sam częsty objaw podrażnienia płata potylicznego, a także halucynacje wzrokowe, ale często pozostaje nierozpoznany. Zwykle pacjenci nie rozróżniają tego objawu osobno w strukturze napadu. Utrata wzroku występuje obustronnie z utratą pól bocznych. Możliwa homonimiczna hemianopia kontralateralna do ogniska ataku. Odczucia pacjentów opisywane są przez nich jako ciemnienie w oczach, „biała ciemność”, zaburzona percepcja kolorów. Być może kurs statusowy z powstaniem tzw. status epilepticus amauroticus.
Napady potyliczne mogą objawiać się objawami autonomicznymi. Należą do nich migrenowy ból głowy, zawroty głowy, nudności i wymioty. Częstym objawem jest ponapadowy ból głowy przypominający migrenę.
Kliniczne objawy drgawek, które występują w ograniczonym zakresie w korze potylicznej, charakteryzują się odchyleniem oczu na bok. Odchylenie oczu można zauważyć wraz z odchyleniem głowy na bok. W większości przypadków obserwuje się odchylenie oczu w kierunku przeciwstronnego ogniska. Jednak opisano przypadki, gdy obserwuje się odwodzenie oczu w kierunku ogniska. Jedną z cech napadów „potylicznych” jest natychmiastowe rozprowadzanie wydzieliny do przednich części mózgu, podczas gdy obraz kliniczny z reguły jest zdominowany przez wyraźną składową motoryczną. Możliwe są drgawki toniczne, toniczno-kloniczne (zarówno hemikonwulsyjne, jak i wtórnie uogólnione). W związku z tym ważne jest, aby zidentyfikować początkowe objawy kliniczne - brak motywacji i nagłe zatrzymanie wzroku, patrzenie na nieistniejące przedmioty, nieuzasadniony uśmiech, objawy wegetatywne i koniecznie udokumentowanie pierwotnej strefy ikonogennej metodą VEM.
Wraz z postępem choroby odnotowuje się oporność padaczki na terapię, często z przebiegiem stanu. Opisano przypadki transformacji w padaczkę Kozhevnikova. Szereg auto-
Rov opisuje możliwość wystąpienia stanu padaczkowego jako pierwszego objawu u pacjentów z MELAS bez wcześniejszych napadów padaczkowych w wywiadzie. Ribacoba R. i in. opisują w swojej publikacji 4 przypadki rozwoju padaczki częściowej ciągłej z ogniskowymi napadami ruchowymi, poprzedzonych epizodami migrenowego bólu głowy w wywiadzie. Miyazaki M. i in. wykazali możliwość kontynuacji ogniskowych mioklonie w obrębie padaczki częściowej ciągłej u pacjentów z MELAS. Araki T. i in. obserwował pacjenta w wieku 37 lat ze stanem padaczkowym napadów ogniskowych w postaci fluktuacji świadomości, homonimicznej hemianopsji w połączeniu z napadowymi epizodami odchylenia oka na bok. W EEG rejestrowano ciągłe wzorce EEG napadów zlokalizowanych w okolicy potylicznej. U dorosłych pacjentów z MELAS przeważają napady ogniskowo-ruchowe, ale w EEG dominuje wieloregionalna aktywność padaczkowata w okolicy potylicznej.
Aktywność padaczkową odnotowuje się w 71% przypadków po wystąpieniu napadów. Badanie elektroencefalograficzne pacjentów z zespołem MELAS charakteryzuje się aktywnością padaczkową w okolicy potylicznej. Wielu autorów wiąże pojawienie się regionalnych zaburzeń padaczkowych z udarami. Według badań Fujimoto S. w ostrym okresie (tj. w ciągu 5 dni po epizodzie udaropodobnym) większość badanych pacjentów z zespołem MELAS miała regionalne fale delta o wysokiej amplitudzie w połączeniu z wielokolcami. Autorzy proponują uznać ten wzorzec za patognomoniczny dla epizodów podobnych do udaru. Poza obszarami potylicznymi, aktywność padaczkowata może rozprzestrzeniać się na obszary skroniowe, obustronnie, a także obustronnie na obszary tylne z rozlanym rozmieszczeniem. Być może pojawienie się reakcji fotoparoksysmalnej podczas rytmicznej fotostymulacji.
Wiodącym znakiem laboratoryjnym jest wzrost poziomu mleczanu we krwi.
ARTYKUŁY ORYGINALNE
wi powyżej 2,0 mmol/l, co prowadzi do rozwoju kwasicy mleczanowej.
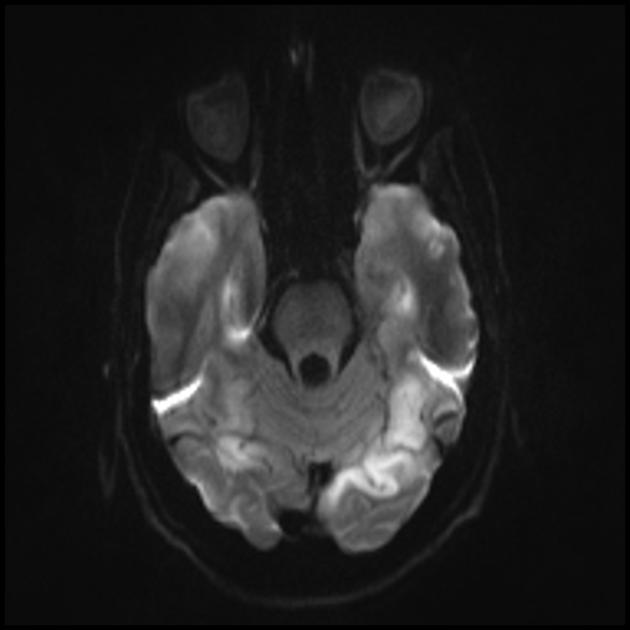
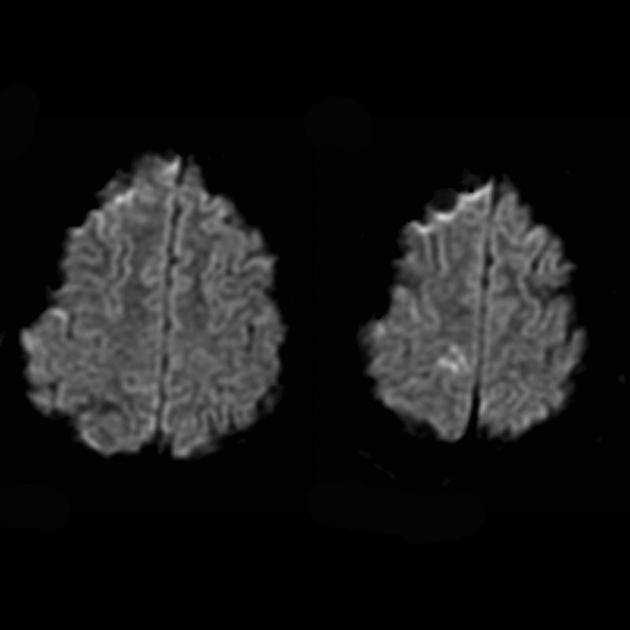
MRI mózgu we wczesnych stadiach choroby może być niezauważalne, nawet jeśli wystąpi padaczka. Metody neuroobrazowania ujawniają strefy zawałowe w półkulach mózgowych (80%), rzadziej w móżdżku i zwojach podstawy mózgu. Może również wystąpić zwapnienie jąder podstawy, zanik kory mózgowej. W badaniu emisji fotonów akumulację izotopu wykrywa się 3-16 dni przed pojawieniem się strefy zawału (spadek sygnału izotopowego) na tomogramie komputerowym mózgu. MRI mózgu pokazuje zmiany zlokalizowane głównie w płatach potylicznych, które mogą być przemijające. Dotknięta jest głównie kora potyliczna, istota biała jest uszkodzona w mniejszym stopniu. Na obrazach T2-zależnych zmiany w mózgu w MELA pojawiają się jako obszary o zwiększonej intensywności sygnału. Wielu autorów kojarzy przejściowo hiperintensywne obszary z odwracalnym obrzękiem naczyniowym.
Angiografia zwykle nie ujawnia nieprawidłowości naczyniowych. MRI ważone dyfuzją wykazuje zmiany związane z obrzękiem naczyniopochodnym.
Histopatologia: Biopsja mięśnia ujawnia włókna z rozdartymi „czerwonymi krawędziami”. Autopsja mózgu charakteryzuje się połączeniem starych i nowych ognisk zawałów, a także atrofią kory z ogniskami martwicy.
Obecnie terapia jest wspomagająca. Głównym kierunkiem leczenia jest poprawa bilansu energetycznego mitochondriów i łańcucha oddechowego. Zastosuj koenzym p10 (80-300 mg/dzień), witaminy K1 i KZ (25 mg/dzień), kwas bursztynowy (do 6 g/dzień), witaminę C (2-4 g/dzień), ryboflawinę (100 mg/dzień dzień) i nikotynamid (do 1 g/dzień). W związku z rozwijającym się wtórnym niedoborem karnityny pacjentom przepisuje się L-karnitynę (do 100 mg/kg/dzień). Witamina E (300-500 mg/dzień) i witamina C (2-4 mg/dzień) są stosowane jako terapia antyoksydacyjna.
Nie ma ogólnie przyjętych schematów leczenia przeciwpadaczkowego MELA. Wielu autorów proponuje wykluczenie leków, które mogą hamować metabolizm energetyczny (barbiturany, leki na kwas walproinowy, a także niektóre leki z innych grup, na przykład chloramfenikol). W piśmiennictwie opisano kilka pojedynczych przypadków nasilenia napadów po zastosowaniu kwasu walproinowego w zespole MELA z mutacją A3243C. Głównymi LPP w leczeniu padaczki w zespole MELA są tegretol (lub trileptal), topamax, keppra w średnich dawkach terapeutycznych. Właściwie dobrana terapia prowadzi do znacznego zmniejszenia częstości napadów drgawkowych wtórnie uogólnionych. Jednak napady z zaburzeniami funkcji wegetatywno-trzewnych i wzrokowych są zwykle oporne na leczenie. W końcowej fazie choroby częstość napadów padaczkowych może się zmniejszyć.
Oto historia przypadku pacjenta z rozpoznaniem zespołu MELAY zweryfikowanego za życia.
Pacjent Ch.A., lat 11, był obserwowany w Centrum Neurologii Dziecięcej i Padaczki. Przy przyjęciu zgłaszano skargi na stopniową utratę zdolności mowy, wyraźne zaburzenie chodu z odmową chodzenia, znaczne pogorszenie widzenia, kapryśność i negatywne zachowanie; codzienne seryjne ataki w postaci drgawek mięśni twarzy, mięśni kończyn górnych i dolnych, a także krótkotrwałe epizody utraty wzroku.
Debiut choroby odnotowano w wieku 5 lat 9 miesięcy. Po raz pierwszy na tle pełnego zdrowia podczas zasypiania pojawił się silny ból głowy, proste halucynacje wzrokowe („żółty promień”), po których nastąpił gwałtowny obrót oczu i głowy na bok oraz rozwój uogólnionego drgawki toniczno-kloniczne, po których odnotowano wymioty. Po 9 miesiącach ataki z tymi samymi objawami powracały i szybko nabierały charakteru seryjnego. Po wyznaczeniu tegretolu w dawce 400 mg na dobę częstotliwość ataków zmniejszyła się do 1 raz na miesiąc. Tegretol został zastąpiony przez Depakine Chrono w dawce 900 mg/dobę, w stosunku do którego obserwowano remisję kliniczną przez 6 miesięcy. Biorąc pod uwagę objaw kliniczny
TOM IV WYDANIE 3 2009
tomatyka, ograniczenie napadów do okresu zasypiania, normalna inteligencja chorego, pozytywna reakcja na walproinian, rozpoznano idiopatyczną padaczkę potyliczną.
W wieku 7 lat powracały ogniskowe napady wersyjne z wtórnym uogólnieniem podczas zasypiania z taką samą częstotliwością 1 raz w miesiącu. Zwiększenie dawki Depakine do 1500 mg/dobę nie spowodowało zmniejszenia częstości napadów. Po dodaniu lamiktalu w dawce 75 mg/dobę napady ustąpiły na 4 miesiące, a następnie powróciły z tą samą częstotliwością. W wieku 8 lat dołączyły ataki z krótkotrwałą utratą wzroku. Od 8 lat 8 miesięcy przed zaśnięciem zaczęły pojawiać się nietypowe nieobecności: gwałtowne mruganie z zamknięciem powiek i założeniem gałek ocznych do góry; świadomość waha się.
W wieku 9 lat pojawiły się wielokrotne, kilkudniowe ataki seryjne z prostymi halucynacjami wzrokowymi w postaci migającego „promienia” przed oczami, z obrotem oczu i głową w prawo. Przed zaśnięciem takie ataki czasami przeradzały się w ogniskowe hemikloniczne, które objawiały się zmniejszeniem twarzy
mięśnie po prawej stronie, drganie głowy w prawo, klony kończyn prawych (większe niż ramię). Czasami po ataku pojawiał się silny ból głowy i wymioty. W tym samym wieku pojawiły się drgawki hamujące: aura w postaci gęsiej skórki na dużym palcu prawej stopy, a następnie krótkotrwałe osłabienie prawej nogi i niezręczność prawej ręki. Do schematu leczenia włączono Topamax w dawce 100 mg/dobę – przez 1 rok nie występowały napady padaczkowe.
Również w wieku 9 lat pojawiły się po raz pierwszy stany napadowe, którym towarzyszył silny ból głowy, wymioty i rozwój prawostronnego niedowładu połowiczego. W niektórych przypadkach takim stanom towarzyszyła amauroza trwająca od kilku minut do kilku dni.
W wieku 10,5 roku ponownie pojawiły się napady w postaci obracania głowy w lewo, szarpanych ruchów gałek ocznych w lewo, trwających do 5 s, częstość do 3 razy na godzinę, codziennie, nawet podczas snu. Dawkę Topamax zwiększono do 150 mg/dobę bez znaczącego efektu. W wieku 10 lat 10 miesięcy. po intensywnym bólu głowy, naprzemiennie
Ryż. 1. Pacjent Ch.A. 10 lat. Diagnoza: zespół MEAE. Objawowa padaczka ogniskowa.
Monitorowanie wideo-EEG (2004): na tle rozproszonego spowolnienia w głównej aktywności mózgu, zarejestrowano ciągłą aktywność padaczkową w lewej okolicy potylicznej. Subkliniczne wzorce EEG napadu zostały również zarejestrowane w lewej okolicy potylicznej z rozprzestrzenieniem się na lewą tylną okolicę skroniową.
Centrum Neurologii Dziecięcej i Padaczki
pod kierunkiem profesora K.Yu. Mukhina zajmuje się diagnostyką i leczeniem zaburzeń lękowych układu nerwowego w Aetei, specjalizuje się w etyckich formach padaczki.
Główne kierunki
zajęcia:
Padaczka u dzieci i młodzieży
Ból głowy
Zaburzenia snu u dzieci
Tiki, moczenie nocne
Badanie dzieci w pierwszych ^ miesiącach życia.
Egzaminy w naszym ośrodku:
Diagnostyka i leczenie chorób układu nerwowego u dzieci
Pełna diagnostyka (w tym przedoperacyjna) i leczenie padaczki
Konsultacje neurologów i epileptologów
Konsultacja pediatry (często chore dzieci, gastroenterologia itp.)
Konsultacja psychiatry i psychologa.
Konsultacja genetyczna z badaniami (w tym kariotypowanie)
Monitoring wideo-EEG (w specjalnie wyposażonych salach Ośrodka lub z wizytą w domu pacjenta)
Elektroencefalografia komputerowa (cyfrowa)
UZDG (dopplerografia ultradźwiękowa) naczyń głowy i szyi
Echoencefalografia (ECHO EG)
Na naszej stronie możesz subskrybować czasopismo „Russian Journal of Child Neurology” przez Internet.
Szczegółowe informacje o pracy Ośrodka w godzinach od 10:00 do 19:00 telefonicznie:
tel.: (+7495) 983-09-03; (+7926) 290-50-30 Tel./Fax: (+7495) 394-82-52
Adres: ul. Borisovskie Prudy, 13 lat, bud. 2. Internet: www.epileptologist.ru E-mail: [e-mail chroniony](dokładna mapa tras na stronie)
TOM IV WYDANIE 3 2009
ogniskowe napady hemkloniczne i wtórnie uogólnione, które stały się seryjne i trwały 48 godzin. Frizium dodano do topamaxu w dawce 10 mg/dzień z chwilowym pozytywnym efektem.
Od 8 roku życia zaczęto zauważać trudności z przyswajaniem materiałów szkolnych; zmniejszona pamięć. Nastąpiło zwiększone zmęczenie, wyczerpanie, zahamowanie aktywności umysłowej. Chłopiec stał się kapryśny, drażliwy, negatywny; tło nastroju zmalało. Od 9 roku życia nastąpił wzrost tej symptomatologii.
Z anamnezy życia wiadomo, że dziecko urodziło się z drugiej prawidłowej ciąży, drugiego porodu w terminie, masa urodzeniowa 2800 g, długość 53 cm Wczesny rozwój psychomotoryczny i mowy był w pełni dostosowany do wieku. Przebyte choroby: ospa wietrzna w wieku 6 lat, częste ostre infekcje wirusowe dróg oddechowych (do 4 razy w roku) od 6 roku życia. Dziedziczność padaczki i innych chorób neurologicznych nie jest obciążona.
W momencie badania (11 lat) stan dziecka był ciężki; reaguje negatywnie na inspekcję. Świadomy, pro-zorientowany
przestrzeń i czas. Bardzo niechętnie nawiązuje kontakt, odmawia wykonania poleceń. Oczopląs samoistny w lewo, głowa pochylona na lewe ramię z skrętem w prawo. Język znajduje się w linii środkowej, odruch gardłowy jest zmniejszony; Obserwuje się dysfagię i dyzartrię. Wizja jest zmniejszona.
Określa się umiarkowaną rozlaną hipotonię mięśniową. Odruchy ścięgien są równomiernie zredukowane. Wystąpił niewielki spadek siły mięśni w kończynach prawych. Nie wykryto patologicznych odruchów stóp. Nie ma obiektywnych danych dotyczących naruszenia wrażliwości. Nie warto w teście Romberga. Odmawia chodzenia. Kiedy próbujesz postawić go na nogi, płacze, siada na podłodze. Brak podczas wykonywania testu wskaźnika palca. Mówi powoli, jednymi słowami, niechętnie.
Dodatkowe metody badania. Monitoring wideo-EEG (2004). Znaczne spowolnienie głównej aktywności nagrywania w tle. W trakcie badania obserwowano ciągłą aktywność padaczkową w lewej okolicy potylicznej z rozprzestrzenieniem się na lewą tylną okolicę skroniową i okresowym tworzeniem wzoru EEG
urodzony w 1993 16.12.05
Ryż. 2. Pacjent Ch.A. 11 lat. Diagnoza: zespół MELAS. Objawowa padaczka ogniskowa.
Monitorowanie wideo-EEG przeprowadzono w dynamice po 1 roku (2005): znaczne spowolnienie aktywności tła mózgu. Podczas rejestrowania snu, w prawym czołowo-centralnym obszarze, którego struktura szczytowa aktywność falowa jest wykrywana w prawym czołowo-centralnym obszarze, rejestrowane jest ciągłe hamowanie regionalne.
ARTYKUŁY ORYGINALNE
stupa (ryc. 1). Określa się również ciągłe hamowanie regionalne w prawym czołowo-centralnym regionie z włączeniem pojedynczych ostrych fal.
Dynamiczne monitorowanie wideo-EEG (2005): Znaczące spowolnienie aktywności mózgu w tle. Badanie odnotowało ciągłe spowolnienie regionalne w prawym regionie czołowo-centralnym. W strukturze deceleracji regionalnej w prawym obszarze czołowo-centralnym ujawnia się aktywność fal szczytowych (ryc. 2).
MRI mózgu. Pierwszy MRI (6 lat) ujawnił pojedynczy sygnał hiperintensywny w trybie T2 w lewej półkuli móżdżku. Badanie MRI w czasie (10,5 roku): ujawniono znaczne pogorszenie pierwotnej zmiany chorobowej wraz z rozprzestrzenianiem się procesu patologicznego szeroko na lewy i prawy region potyliczno-ciemieniowy obu półkul mózgu (profesor A.A. Alikhanov).
Wzrokowe potencjały wywołane: znaczące zmiany morfologiczne i czynnościowe w wzrokowym układzie doprowadzającym na poziomie nerwu wzrokowego i części korowej analizatora wzrokowego, bardziej wyraźne po lewej stronie.
Konsultacja okulisty: częściowy zanik nerwów wzrokowych. Elementy agnozji korowej.
Elektrokardiogram: rytm ektopowy z przyspieszeniem do 100 uderzeń na minutę.
Pionowa pozycja elektrycznej osi serca. Zmiany w procesach repolaryzacji, które są bardziej wyraźne w ortostazie.
Elektroneuromiografia: ujawniła pierwotny typ zmiany mięśniowej. Prędkości przewodzenia wzdłuż nerwów obwodowych nie ulegają zmniejszeniu.
Badanie poziomu mleczanu we krwi: zawartość mleczanu we krwi wynosi 3,0 mmol / l (norma do 1,8).
Biorąc pod uwagę obecność napadów padaczkowych pochodzących z okolic potylicznych kory mózgowej, opornych na terapię, epizodów udaropodobnych, okresów ślepoty, osłabienia funkcji poznawczych, obecności sygnałów hiperintensywnych w móżdżku i tylnych obszarach kory mózgowej na MRI, wzrost poziomu mleczanu we krwi, pacjentka miała rozpoznanie zespołu MELAS. Podczas badania genetycznego w krwinkach stwierdzono mutację A3243G w stanie heteroplazmatycznym (diagnoza została przeprowadzona w Państwowym Centrum Badawczym Rosyjskiej Akademii Nauk Medycznych), a diagnoza została zweryfikowana.
Obserwacja w obserwacji wykazała szybki postęp naruszeń wyższych funkcji psychicznych, rozwój ślepoty korowej, całkowite unieruchomienie pacjenta, a następnie początek zgonu w wieku 12 lat i 10 miesięcy. (po 7 latach od wystąpienia choroby).
Bibliografia
1. Nikołajewa E.A., Temin P.A. Choroby mitochondrialne, którym towarzyszy upośledzony rozwój neuropsychiczny. Zespół MELAS // Dziedziczne zaburzenia rozwoju neuropsychicznego dzieci. Poradnik dla lekarzy pod redakcją Temina P.A. Kazantseva L.Z. - Medycyna, 2001. - S. 96-107.
2. Nikolaeva E.A., Temin P.A., Nikanorova M.Yu., Klembovsky A.I., Sukhorukov V.S., Dorofeeva M.Yu., Korsunsky A.A. Leczenie dziecka z zespołem mitochondrialnym MELAS (encefalopatia mitochondrialna, kwasica mleczanowa, epizody udaropodobne) // Rosyjski Biuletyn Perinatologii i Pediatrii. - 1997. - nr 2. - S. 30-34.
3. Smirnova I.N., Kistenev B.A., Krotenkova M.V., Suslina ZA. Udarowy przebieg encefalomiopatii mitochondrialnej (zespół MELAS) // Atmosfera. Choroby nerwowe. - 2006r. - nr 1. - S. 43-48.
4. Temin PA, Nikanorova M.Yu., Nikolaeva E.A. Zespół MELAS (encefalomiopatia mitochondrialna, kwasica mleczanowa, epizody podobne do udaru): główne objawy, kryteria diagnostyczne, opcje leczenia // Nevrol. czasopismo - 1998. - nr 2. - S. 43-48.
5. Ajmone-Marsan C., Ralston B. Napad padaczkowy, jego morfologia czynnościowa i znaczenie diagnostyczne. - Springfield (IL): Charles C. Thomas, 1957. - P. 3-231.
6. Aldrich M.S., Vanderzant C.W., Alessi A.G., Abou-Khalil B., Sackellares J.C. Napadowa ślepota korowa z trwałą utratą wzroku // Padaczka. - 1989. - V. 30. - P. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. Przypadek MELAS z złożonym stanem częściowym padaczkowym // Rinsho Shinkeigaku. - 2001. - V. 41(8). - str. 487-90.
TOM IV WYDANIE 3 2009
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptic fenotypy związane z zaburzeniami mitochondrialnymi // Neurologia. - 2001. - V. 56(10). - str. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. Kwas walproinowy zaostrza padaczkę z powodu MELAS u pacjenta z mutacją A3243G mitochondrialnego DNA // Metab Brain Dis. - 2007 - V. 22(1). - str. 105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. i in. Patologia molekularna MELAS i MERRF. Związek między obciążeniem mutacją a fenotypami klinicznymi // Mózg. - 1997. - V.120. - str. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Polimorfizm padaczki związany z mutacją A3243G mitochondrialnego DNA (MELAS): przyczyny opóźnionej diagnozy // Rev Neurol (Paryż). - 2004. - V. 160(8-9). - str. 824-829.
12. Dvorkin G., Andermann F., Carpenter S. Migrena klasyczna, nieuleczalna padaczka i udary mnogie: zespół związany z encefalopatią mitochondrialną / W: Andermann F., Lugaresi E., red.. migrena i epilepsja. - Boston: Butterworths, 1987. - P. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Wyniki seryjnych elektroencefalografów u pacjentów z MELAS // Pediatr Neurol. - 1999. - V. 20(1). - str. 43-48.
14. Goto Y., Nonaka I., Horai SA. Mutacja w genie tRNA leu (UUR) związana z podgrupą encefalomiopatii mitochondrialnych MELAS // Natura. - 1990. - V. 348. - P. 651-653.
15. Hasuo K., Tamura S., Yasumori K., Uchino A., Goda S., Ishimoto S. i in. Tomografia komputerowa i angiografia w MELAS (miopatia mitochondrialna, encefalopatia, kwasica mleczanowa i epizody udaropodobne): opis 3 przypadków // Neuroradiologia. - 1987.-V. 29. - str. 393-397.
16. Hirano M., Pavlakis S.G. Miopatia mitochondrialna, encefalopatia, kwasica mleczanowa i epizody udarowe (MELAS): Aktualne koncepcje // J. clin. Neurol. - 1994. - V. 9. - P. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto, Hirose G. Napady padaczkowe u pacjenta z miopatią mitochondrialną, encefalopatią, kwasem mlekowym i epizodami udaropodobnymi ( MELAS) // Jpn J Psychiatry Neurol. - 1989. - V. 43(3). - str. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J. i in. Encefalomiopatia mitochondrialna z podwyższeniem poziomu mleczanu i pirogronianu i zawałami mózgu // Neurologia. - 1984. - V. 34. - P. 72-77.
19. Kuzniecky R. Objawowa padaczka płata potylicznego // Padaczka. - 1998. - V. 39 Suppl 4. - P. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Głębokość i bezpośredni zapis korowy w zaburzeniach napadowych pochodzenia pozaskroniowego // Neurologia. - 1976. - V. 26. - P. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Kliniczne wzorce napadów u pacjentów z padaczką z potylicznymi ogniskami elektroencefalograficznymi // Neurologia. - 1975. - V. 25. - P. 463-471.
22. Matthews P.M., Tampieri D., Berkovic S.F., Andermann F., Silver K., Chityat D. i in. Rezonans magnetyczny wykazuje specyficzne nieprawidłowości w zespole MELAS // Neurologia. - 1991. - V. 41. - P. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. Przypadek MELAS związany z epilepsją częściową ciągłą // No To Hattatsu. - 1991. - V. 23(1). - str. 65-70.
24. Montagna P., Gallassi R., Medori R., Govoni E., Zeviani M., Di Mauro S. i in. Zespół MELAS: charakterystyczne cechy migrenowe i padaczkowe oraz transmisja matczyna // Neurologia. - 1988. - V. 38. - P. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N. i in. Mózgowy przepływ krwi w miopatii mitochondrialnej, encefalopatii, kwasicy mleczanowej i epizodach udarowych // Udar. - 1993. - V. 24. - P. 304-309.
26. Pavlakis S.G., Phillips PC, Di Mauro S. et al. Miopatia mitochondrialna, encefalopatia, kwasica mleczanowa i epizody podobne do udaru: charakterystyczny zespół kliniczny // Neurol. - 1984. - V. 16. - P. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. Charakterystyka stanu padaczkowego w MELAS. Analiza czterech przypadków // Neurologia. - 2006. - V. 21(1). - str. 1-11.
28. Williamson PD, Spencer S.S. Cechy kliniczne i EEG złożonych napadów częściowych pochodzenia pozaskroniowego // Padaczka. - 1986. - V. 27 (Suppl 2). - str. 46-63.
29. Williamson P.D., Thadani V.M., Darcey T.M., Spencer D.D., Spencer S.S., Mattson R.H. Padaczka płata potylicznego: charakterystyka kliniczna, wzorce rozprzestrzeniania się napadów i wyniki operacji // Ann Neurol. - 1992. - V. 31. - S. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Paradoksalne działanie walproinianu sodu, który nasila padaczkę MELAS u pacjenta z mutacją A3243G w mitochondrialnym DNA // Central European Journal of Medicine. - 2007. - V. 2(1). - P.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. Obrzęk naczyniowy na MELAS: seryjne badanie z obrazowaniem MR ważonym dyfuzją // Neurologia. - 1999. - V. 53. - P. 2182-2184.
Zespół MELAS (MELAS) został po raz pierwszy opisany w 1984 roku przez S. Pavlakisa i współpracowników. Ale niektórzy badacze uważają, że syndrom został już wcześniej określony przez takie pojęcia, jak rodzinna poliodystrofia, kwasica mleczanowa.
Esencja patologii
W 1994 roku S. Pavlakis i Mizio Hirano opisali 110 przypadków choroby. MELAS (mitochondrialna encefalomiopatia, kwasica mleczanowa i epizody podobne do udaru) to wieloukładowa postępująca choroba neurodegeneracyjna. Jest polimorficzny i charakteryzuje się encefalopatią z drgawkami i otępieniem, kwasicą mleczanową. Choroba jest spowodowana mutacjami w mitochondrialnym DNA (mtDNA). Ta choroba ma inną nazwę - encefalomiopatia mitochondrialna.
Informacje ogólne
Od 25 do 44% przypadków choroby ma charakter dziedziczny, przenoszony przez linię matczyną. W innych przypadkach pojawia się po raz pierwszy. Obecnie znanych jest ponad 10 genów, które mutują i prowadzą do rozwoju tego zespołu. Są to geny kodujące funkcje transferu RNA. Zespół MELAS odnosi się do chorób mitochondrialnych (MD) z nieprawidłową akumulacją mitochondriów, co skutkuje zaburzeniem całego systemu metabolizmu energetycznego komórki.
Choroby z tej grupy są przenoszone tylko przez linię matczyną. Dzięki nim najbardziej zależne od energii narządy i tkanki są dotknięte różnymi kombinacjami: mięśniami szkieletowymi, sercem, mózgiem, wzrokiem, wątrobą i nerkami.
Objawy są polimorficzne i mogą wystąpić w każdym wieku. Obejmuje objawy cukrzycy, utratę słuchu, drgawki, endokrynopatie, niski wzrost, patologie serca, całkowitą niezdolność do ćwiczeń i zaburzenia psychomotoryczne.
Wcześniej rozwój psychomotoryczny był całkowicie normalny. Nie zidentyfikowano pacjentów z takimi samymi objawami, ponieważ mutacja dotyczy wielu genów: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTTS2, MTND1, 5, 6. Ich liczba stale rośnie.
U 80% pacjentów zespół MELAS jest spowodowany substytucją punktową A3243G w genie tRNA leucyny (UUR).
Częstotliwość została zbadana w sposób niewiarygodny. Jest tylko kilka danych: np. w Finlandii częstość mutacji A3243G wynosiła 16:100 tys. populacji; w Anglii - 1 przypadek na 13 tys. osób.
Zmiany patologiczne

Charakterystyczną cechą patologiczną zespołu MELAS są postrzępione czerwone włókna (RRF), które można zaobserwować w tkance mięśniowej ze specjalnym tricolorem Gomory. Są wynikiem zmutowanych genów i morfologicznego substratu uszkodzenia mtDNA, powstałego w wyniku proliferacji tych nieprawidłowych mitochondriów.
Czym właściwie jest mitochondrium
Mitochondria to dwubłonowe organelle komórki eukariotycznej (komórki posiadającej jądro), której główną funkcją jest dostarczanie energii. Oznacza to, że w rzeczywistości mitochondria są bazą energetyczną komórek, ich stacjami energetycznymi.
Liczba mitochondriów w komórkach może wahać się przez całe życie od kilku do tysięcy. A więcej z nich dzieje się w komórkach związanych z produkcją energii.
Same mitochondria są najczęściej okrągło-wydłużone, o wielkości od 1 do 10 mikronów. Mogą zamarzać bez ruchu lub poruszać się wewnątrz cytoplazmy komórki. Zwykle przenoszą się tam, gdzie potrzeba więcej energii.
Na wewnętrznej błonie mitochondriów znajdują się narośla (cristae), na których znajdują się całe układy enzymów. Zasadniczo są to związki białkowe. Liczba cristae zależy od intensywności procesów syntezy. Na przykład w mitochondriach komórek mięśniowych jest ich zawsze dużo.
Mitochondria posiadają autonomiczny system syntezy białek – DNA, RNA i rybosomy. Część niezbędnych białek jest syntetyzowana przez same mitochondria - 5%, a część pozyskiwana jest z cytoplazmy - 95%. Energia jest pozyskiwana ze związków organicznych poprzez różne reakcje enzymatyczne.
Niektóre z tych reakcji zachodzą z udziałem tlenu, tj. zachodzi utlenianie, a po innych uwalniany jest CO 2 z przeniesieniem protonów wodoru i uwolnieniem energii. Innymi słowy, mitochondrium jest aktywnym uczestnikiem oddychania komórkowego.
Reakcje te zachodzą na grzebieniach lub w samych mitochondriach, co jest tak ważne dla komórki, że wyleczona komórka będzie całkowicie zdrowa.
Patogeneza

Na pierwszy rzut oka w zespole MELAS stan przypomina wariant po udarze. Ale w rzeczywistości jest nietypowy: występuje u młodych ludzi, często wywoływany przez choroby zakaźne i może występować w postaci złośliwego bólu głowy przypominającego migrenę, drgawek.
Angiografia nie daje żadnych patologii naczyniowych. Mogą występować normalne naczynia lub zwiększone kalibry niektórych tętnic, żył lub występuje przekrwienie naczyń włosowatych.
MRI pokazuje, że ostre uszkodzenie mózgu w zespole MELAS może migrować, a nawet zanikać. Niektóre ogniska ulegają wahaniom. Dla typowego udaru jest to zupełnie nietypowe.
W zespole MELAS występuje wieloogniskowa martwica. W większości jest to zauważalne w części potylicznej (lokalizacja tylna) kory mózgowej i istoty białej podkory. Ale mogą również wystąpić w innych częściach mózgu. Obszary te przypominają martwicę w zawale serca, ale znajdują się poza basenami centralnych naczyń mózgowych.
Objawy objawowe

Zwykle zespół MELAS u dzieci debiutuje w wieku 6-10 lat (może rozpocząć się w wieku 3 lat i 40 lat). Wczesny początek choroby jest bardziej typowy i dotyczy 90% pacjentów. Z wczesnym początkiem choroba przebiega trudniej. Pacjenci są zwykle niewymiarowi, słabi umięśnieni i absolutnie nieprzystosowani do wysiłku fizycznego.
Każde napięcie lub aktywność fizyczna powodują, że czujesz się gorzej. Spośród narządów wewnętrznych serce jest dotknięte niedożywieniem mięśni i przewodzenia, a następnie rozwija się niewydolność sercowo-naczyniowa. Występują również nefropatia, cukrzyca, zaburzenia żołądkowo-jelitowe z wymiotami, osłabienie słuchu. Charakteryzuje się bólem mięśni, brakiem odruchów, niedowładem, drgawkami, IPE, utratą przytomności. Osłabienie mięśni (zespół miopatyczny) i niedosłuch odbiorczy są również typowe dla tej patologii.
Endokrynopatia jest reprezentowana nie tylko przez cukrzycę, ale także przez niedobór hormonu wzrostu. Zaburzenia serca i nerek są rzadkie w rozwoju omawianej choroby.
Napady padaczkowe w zespole MELAS są bardzo zmienne. Mogą być ogniskowe, uogólnione, toniczno-kloniczne i miokloniczne. Charakterystyczna jest bezwzględna niewrażliwość napadów na leczenie przeciwdrgawkowe. Często zdarza się, że lekarze diagnozują epilepsję i przepisują np. kwas walproinowy. Po nim stan zdrowia gwałtownie się pogarsza, a drgawki nasilają się, ponieważ powoduje to depresję mitochondriów. Chociaż rozwija się demencja, rzadko staje się ona objawem jawnym.
Jest również charakterystyczny dla choroby, ale występuje również w wielu innych patologiach, dlatego nie może służyć jako podstawa diagnozy. Dopiero w połączeniu z migrenami, drgawkami i/lub zjawiskami podobnymi do udaru można podejrzewać wystąpienie zespołu MELAS. Nawet tak rozległe objawy nie dają prawidłowej diagnozy. Postęp procesu przebiega na różne sposoby.
oznaki

Charakterystyczną cechą kliniczną zespołu MELAS są epizody udaropodobne (IPE), w których nagle pojawiają się objawy neurologiczne. IPE charakteryzuje się asymetrią zmian. Mogą być wielokrotne.
Selektywność takiej lokalizacji daje również pewne ogniskowe objawy:
- hemianopsja (ślepota korowa);
- niedowład połowiczy;
- afazja sensoryczna (niezrozumienie słów);
- akalkulia (naruszenia konta);
- agrafia (naruszenia pisowni);
- ataksja (upośledzona koordynacja ruchów dobrowolnych);
- zmiany w świadomości.
Często zdarza się, że objawy podobne do udaru powracają co 1 do 3 miesięcy. Osobliwością ostrych epizodów w MELAS jest to, że mają one szybką regresję, ale często nawracają, to znaczy przechodzą bez śladu. Ponadto u pacjentów z tą chorobą zwapnienia odkładają się w jądrach podstawy (znajduje się to w CT).
Epizody przypominające udar mózgu często rozwijają się w wieku 5-15 lat. Nigdy nie stają się wynikiem choroby zakrzepowo-zatorowej. Angiopatia w MELAS jest spowodowana hiperproliferacją tych samych mitochondriów.
IPE w objawach objawia się nawracającymi atakami bólu głowy, zawrotami głowy, niedowładem, paraliżem kończyn, nerwów czaszkowych. Mężczyzna jest całkowicie zdemoralizowany.
Kwasica mleczanowa w zespole MELAS

Jego głównym winowajcą jest nadmiar kwasu mlekowego we krwi i tkankach układu nerwowego. To znacznie zmniejsza kwasowość krwi w tętnicach. Taka kwasica jest częstym towarzyszem cukrzycy, która występuje w zespole MELAS.
Na wczesnym etapie objawy są niespecyficzne. Obserwuje się następujące objawy: ogólne osłabienie, ból w klatce piersiowej, apatia, senność. Bardzo charakterystyczne są bóle mięśni po wysiłku fizycznym i przerywany, szybki oddech bez zapachu.
W środkowej fazie gromadzi się kwas mlekowy i dochodzi do zespołu hiperwentylacji (HVS). Dwutlenek węgla gromadzi się we krwi. Zaczyna się tworzyć głośny oddech - Kussmaul. Ciśnienie spada, by zapaść, pojawia się skąpomocz. Pacjent staje się niespokojny, majaczy, a następnie wraz z rozwojem śpiączki traci przytomność - to ostatni etap. Objawy kwasicy mleczanowej rozwijają się szybko, a mianowicie w ciągu kilku godzin. Potem przychodzi śmierć.
Środki diagnostyczne

Jak już wspomniano, ze względu na polimorfizm objawów oraz mutację dużej liczby genów, rozpoznanie zespołu MELAS jest trudne. Trzymany:
- ogólne i biochemiczne badania krwi;
- biopsja mięśnia;
- badanie genetyczne z analizą porównawczą wśród chorych krewnych;
- TK mózgu: obszary zawałów częściej w półkulach, rzadziej w móżdżku, zwojach podstawy mózgu;
- wzrost kalibru naczyń krwionośnych (tętnic, żył, naczyń włosowatych);
- Diagnostyka DNA: poszukiwanie charakterystycznych mutacji punktowych w mtDNA.
Metody terapii
Leczenie zespołu MELAS nie zostało opracowane i obecnie jest nieuleczalne. Są tylko próby spowolnienia procesu pokonania. Leczenie przebiega dwukierunkowo: terapia postsyndromiczna (padaczka, cukrzyca) oraz patogenetyczna. Jednak obecnie nie ma skutecznej terapii patogenetycznej.
Istnieją leczenie objawowe: przy ubytku słuchu aktywnie stosuje się aparaty słuchowe, przy osłabieniu mięśni oddechowych prowadzi się terapię oddechową. Zauważono, że przy zespole MELAS we krwi pacjenta poziom L-argininy jest znacznie obniżony podczas IPE. Dlatego terapię prowadzi się preparatami argininy i witaminami. Badany jest pozytywny wpływ koenzymu Q lub idebinonu (noben), preparatów kwasu bursztynowego, witamin K 1 i K 3, B 2 , B 3 , E, C; L-karnityna, antyoksydanty (mexidol, mildronian), korektory kwasicy mleczanowej (dimefosfon) - wszystkie poprawiają metabolizm energetyczny komórki. W leczeniu napadów walproiniany i barbiturany nie są przepisywane, ponieważ hamują mitochondria.
W celu zapobiegania temu syndromowi najlepiej zastosować metodę IVF. Jeśli kobieta wie, że ma przypadek manifestacji tej choroby w swojej rodzinie, cytoplazmę do zapłodnienia pobiera się od zdrowej kobiety. Metoda jest jeszcze na etapie badań, nie jest masowa.
Niektóre funkcje
Rozpoznanie zaburzeń mitochondrialnych wymaga bardzo ostrożnego podejścia do terapii. Musi być w nim koniecznie uwzględnione środki działania metabolicznego. Stabilizują procesy oddychania tkankowego, fosforylacji oksydacyjnej w komórkach. Tylko systematyczne wdrażanie takiego leczenia może pomóc w utrzymaniu stanu pacjentów, zapobiegając epizodom udarów.
Prognoza
Rokowanie jest niekorzystne ze względu na brak skutecznego leczenia. Średnia długość życia od pierwszego wystąpienia objawów zwykle nie przekracza pięciu lat. Brak wiedzy o przyczynach choroby prowadzi do tego, że nie znaleziono jeszcze optymalnego schematu leczenia. Wszystko to sprawia, że szanse na wyleczenie są minimalne.
Zespół MELAS to zaburzenie mitochondrialne charakteryzujące się zajęciem mięśni i OUN.
MELAS (ang. Encefalomiopatia mitochondrialna, kwasica mleczanowa i epizody udaropodobne - „encefalomiopatia mitochondrialna, kwasica mleczanowa, epizody podobne do udaru”) to postępująca choroba neurodegeneracyjna charakteryzująca się wymienionymi w tytule objawami, której towarzyszą objawy polimorficzne - udar, cukrzyca, drgawki, zmniejszenie utraty słuchu, choroby serca, niski wzrost, endokrynopatie, nietolerancja wysiłku i zaburzenia neuropsychiatryczne.
Fabuła.
Zespół MELAS został po raz pierwszy opisany w 1984 roku przez Pavlakisa i współpracowników; dziesięć lat później Pavlakis i Mizio Hirano opublikowali przegląd 110 spraw.
typ dziedziczenia:
macierzyński
Epidemiologia:
Dokładna częstotliwość choroby nie jest znana. W piśmiennictwie niewiele jest danych dotyczących występowania choroby. W północnej Finlandii częstość mutacji A3243G wynosi 16,3: 100 000.
Patogeneza:
Mutacjom mitochondrialnego DNA, które kontrolują łańcuch oddechowy mitochondriów, towarzyszy zakłócenie procesów fosforylacji oksydacyjnej, najważniejszego źródła energii dla procesów metabolicznych w komórce.
Objawy kliniczne
W wieku 40 lat pacjenci z MELAS przyjmowani są do poradni przemijającego napadu niedokrwiennego, a także z padaczką, nawracającymi wymiotami, bólem głowy i osłabieniem mięśni. U tych pacjentów często stwierdza się klinicznie demencję.
Młody wiek i brak czynników ryzyka charakterystycznych dla udaru mózgu sprawiają, że MELAS jest bardziej przemyślany.
Dane laboratoryjne
Kwasica mleczanowa - podwyższony poziom mleczanu i pirogronianu.
Dane wizualizacji
Zmiany w mózgu są podobne do zmian w udarze.
Różnice w stosunku do udaru
1) dotknięte obszary nie pokrywają się z granicami naczyń tętniczych.
2) przy powtarzających się atakach ogniska są wizualizowane w innej lokalizacji.
+ dane kliniczne (młody wiek, brak czynników ryzyka udaru).
CT
Wiele obszarów hipodensyjnych niezgodnych z łożyskiem naczyniowym.
Zwapnienie jąder podstawy (najczęściej u starszych pacjentów).
Zanik występuje na tle regresji i poprawy klinicznej.
MRI
Ostry zawał
Do różnicowania z udarem stosuje się ADC i DWI (ograniczenie dyfuzji w udarze (obrzęk cytotoksyczny), a w MELAS dyfuzja jest nieznacznie ograniczona lub niezmieniona (obrzęk wazogenny).
Zaangażowanie w patologiczny proces podkorowej istoty białej mózgu.
Pogorszenie wizualizacji klarowności konturów zwojów i wzrost sygnału z nich na obrazach T2-ważonych.
Przewlekły zawał
Zmiany mogą być symetryczne lub asymetryczne.
Zanik ogniskowy występuje na tle regresji i poprawy klinicznej.
Najczęściej atakowane są płaty ciemieniowe, potyliczne i skroniowe mózgu.
spektroskopia MR
Zwiększony poziom mleczanu.




Materiały przeznaczone są dla neurologów, terapeutów i lekarzy rodzinnych.
Sergey Lichaczow, kierownik, MD. nauk ścisłych, profesor;
Inessa Pleshko, wiodący badacz, dr inż. Nauki, Oddział Neurologiczny Republikańskiego Centrum Naukowo-Praktycznego Neurologii i Neurochirurgii.
Autosomalna dominująca arteriopatia mózgowa z zawałami podkorowymi i leukoencefalopatią (CADASIL) jest postępującą chorobą autosomalną dominującą, której objawy kliniczne obejmują nawracające podkorowe udary niedokrwienne, migrenę, otępienie podkorowe i zaburzenia afektywne. Obecna częstość występowania – 1 przypadek
na 100 000 mieszkańców.
Republikańskie Centrum Naukowo-Praktyczne Neurologii i Neurochirurgii przyjmuje 7 pacjentów (w tym 4 kobiety) z CADASIL; wiek - od 32 do 68 lat. Zbadano je neurologicznymi, molekularnymi metodami genetycznymi. Były charakterystyczne objawy; w historii - migrena, nawracające udary lakunarne i zaburzenia afektywne. MRI mózgu ujawniło charakterystyczne dla CADASIL zawały podkorowe i leukoencefalopatię.
W wyniku diagnostyki genetyki molekularnej 2 osoby miały heterozygotyczną mutację w genie Notch3 na 19 chromosomie, która powoduje CADASIL. Geny Notch kodują receptory transbłonowe zaangażowane w ontogenezę komórki. Za pomocą CADASIL w większości przypadków określane są mutacje zmiany sensu, przez co zmienia się struktura białka transbłonowego i jego funkcje są upośledzone.
Patogeneza CADASIL nie jest do końca jasna. Uważa się, że głównym czynnikiem jest arteriopatia z postępującą niedrożnością małych naczyń perforujących istotę białą mózgu (prowadzącą do przewlekłej hipoperfuzji). Jednocześnie występują charakterystyczne ziarniste wtrącenia osmofilne, powodujące proliferację składników błony podstawnej, pogrubienie błony środkowej i mechaniczne uciskanie małych tętnic. W efekcie dochodzi do uszkodzenia bariery krew-mózg – rozwija się obrzęk.
Dodatkowym czynnikiem patologicznym jest aktywacja astrocytów w pobliżu ściany naczynia. Uwalniają śródbłonek-1, powodując zwężenie naczyń i upośledzenie przepływu krwi.
Skład ziarnistych wtrąceń osmofilnych jest nieznany. Przyjmuje się, że jednym z ich składników jest białko Notch3. W biopsjach skóry pacjentów z mutacją Notch3 ziarnistości osmofilne i zwyrodnienie komórek mięśni gładkich można wykryć nawet przed ukończeniem 20. roku życia.
Diagnostyka kliniczna CADASIL:
- obciążona historia rodzinna;
- rozwój pierwszych objawów choroby przed 50 rokiem życia;
- obecność dwóch z następujących objawów - migrena, nawracające udary, zaburzenia nastroju, otępienie podkorowe.
Należy wykluczyć naczyniowe czynniki ryzyka związane etiologicznie z objawami neurologicznymi. MRI pokazuje uszkodzenie istoty białej półkul mózgowych i brak zawałów korowych.
Wiarygodną diagnozę „CADASIL” potwierdza pozytywny wynik molekularnej diagnostyki genetycznej lub wykrycie arteriopatii z charakterystycznymi ziarnistymi wtrąceniami osmiofilowymi w biopsji skóry lub mięśni.
Najczęstszymi objawami CADASIL są przemijające ataki niedokrwienne i udary niedokrwienne, obserwowane u prawie 85% pacjentów.
Charakteryzują się nawrotowym przebiegiem, objawiającym się klasycznymi zespołami udarów lakunarnych i całkowitą remisją kliniczną po kilku dniach lub tygodniach.
Drugim najczęstszym są zaburzenia funkcji poznawczych (odnotowane u 60% pacjentów). Może rozpocząć się w wieku 35 lat, czasami nawet przed epizodami niedokrwiennymi. Około 75% pacjentów CADASIL rozwija demencję. Pierwszym objawem jest zwykle migrena; często występuje przed 20 rokiem życia i zwykle poprzedza udar.
Dane dotyczące zaangażowania serca w proces patologiczny w CADASIL są sprzeczne. L. Oberstein i in. (2003) stwierdzili, że 25% pacjentów, u których zdiagnozowano CADASIL, miało historię ostrego zawału mięśnia sercowego lub patologii załamka Q na elektrokardiogramie. W innym badaniu Cumurciuc i in. (2006) nie stwierdzili pozytywnej historii sercowej u 23 osób z mutacją Notch3.
Objawy kliniczne CADASIL i mikroangiopatii mózgowej o różnej etiologii są podobne – wymagana jest diagnostyka różnicowa.
Aby na czas określić CADASIL u pacjentów i ich rodzin, konieczne jest odwołanie się do molekularnych metod genetycznych i / lub badań histologicznych.
Zespół MELAS
Encefalomiopatia mitochondrialna z kwasicą mleczanową i epizodami udaropodobnymi (MELAS) jest rzadką chorobą dziedziczną spowodowaną patologią genomu mitochondrialnego, zaburzonym metabolizmem energetycznym oraz funkcjonowaniem najbardziej zależnych od energii narządów i tkanek (OUN, mięśnie sercowe i szkieletowe, oczy, nerki, wątroba, szpik kostny, układ hormonalny). Duża zmienność objawów klinicznych zespołu MELAS oraz rzadkie występowanie predestynują lekarza do trudności diagnostycznych.
W Republikańskim Centrum Naukowo-Praktycznym Neurologii i Neurochirurgii obserwuje się 3 pacjentów (46-letnia kobieta i jej synowie w wieku 24 i 23 lat) z rozpoznanym zespołem MELAS. Przeszli badania kliniczne i neurologiczne, molekularną diagnostykę genetyczną, rezonans magnetyczny mózgu.
Wszystkie są krótkie; w historii - objawy patologii mitochondrialnej: niedosłuch czuciowo-nerwowy, migrenowe bóle głowy, słaba tolerancja wysiłku. Debiut choroby to uogólnione napady drgawkowe. U 2 pacjentów pierwsze objawy pojawiły się przed 20 rokiem życia; następowały po sobie napady padaczkowe, epizody zaburzeń widzenia z obecnością ognisk w neuroobrazowaniu w okolicy potylicznej i skroniowej, wzrost poziomu mleczanów we krwi i płynie mózgowo-rdzeniowym. 1 osoba miała umiarkowany spadek funkcji poznawczych; według USG serca - kardiomiopatia przerostowa; cukrzyca.
Badanie genetyki molekularnej ujawniło wieloukładowe zmiany typowe dla MELAS, dużą zmienność i różny stopień objawów klinicznych, odpowiadające liczbie kopii mutanta A3243G w genie tRNA Leu(UUR).
MELAS charakteryzuje się matczynym typem dziedziczenia, występowaniem sporadycznych przypadków wystąpienia mutacji de novo; akumulacja w komórkach – zarówno normalnych, jak i zmutowanych – mitochondrialnego DNA (heteroplazmia) oraz losowa dystrybucja podczas podziału między komórki potomne (segregacja mitotyczna). Na poziomie genetycznym przyczyną zespołu MELAS jest heteroplazmatyczna rearanżacja 3243A>G w genie tRNALeu(UUR) (wykryto 80% przypadków).
Patogeneza choroby nie została jeszcze zbadana. Istnieją 2 główne teorie - „angiopatia mitochondrialna” i „cytopatia mitochondrialna”. Wiadomo, że zmiana udaropodobna nie odpowiada strefom naczyniowym i rozciąga się na otaczające obszary z powodu współistniejącego obrzęku naczyniopochodnego spowodowanego przedłużoną aktywnością padaczkową. Jak sugerowano, epizody podobne do udaru są spowodowane nadpobudliwością nerwową w ograniczonym obszarze mózgu. Powstaje z dysfunkcji mitochondriów w komórkach śródbłonka naczyń włosowatych, neuronach lub astrocytach; depolaryzuje sąsiednie neurony, prowadząc do rozprzestrzeniania się aktywności epileptycznej.
Ponadto w przerwach między epizodami udaropodobnymi, według tomografii emisyjnej pojedynczych fotonów (SPECT), pacjenci z MELAS mają hipoperfuzję kory tylnego zakrętu obręczy, co wskazuje na zaburzenie hemodynamiki mózgowej.
Naruszenie fosforylacji oksydacyjnej, przerwanie mitochondrialnego łańcucha oddechowego przyczyniają się do dominacji metabolizmu katabolicznego i zmian z cyklu Krebsa na glikozę beztlenową z akumulacją mleczanu. Wysoki poziom tego ostatniego w OUN zwykle koreluje z okresami objawów neurologicznych.
Głównymi objawami klinicznymi MELAS są epizody podobne do udarów, kwasica mleczanowa i obecność „rozerwanych czerwonych włókien” w próbkach z biopsji mięśni. Dodatkowymi objawami mogą być otępienie, psychoza, napady padaczkowe, migrenowe bóle głowy, ataksja, miopatia, zwapnienie jąder podstawy w obrazowaniu neuroobrazowym, zanik optyczny, retinopatia, głuchota, cukrzyca, pseudoniedrożność jelit, kardiomiopatia.
Wczesny wiek debiutu MELAS wynosi od 5 do 20 lat, istnieją jednak obserwacje późnego zachorowania – w 5.–6. dekadzie życia. Zdarzają się przypadki, gdy zespół zaczął się po zaburzeniach kardiologicznych.
Wieloukładowy charakter zmian w MELAS komplikuje rozpoznanie kliniczne.
Dziedziczny charakter choroby zobowiązuje do przeprowadzenia molekularnych badań genetycznych w celu postawienia trafnej diagnozy.
i zidentyfikować innych pacjentów - spośród krewnych pacjenta.
Materiały przeznaczone są dla neurologów, terapeutów i lekarzy rodzinnych.
| Miopatia mitochondrialna, encefalomiopatia, kwasica mleczanowa i epizody udaropodobne | |
|---|---|
| Zwapnienie jąder podstawy, zanik móżdżku, zwiększony poziom mleczanu; Obraz TK osoby, u której zdiagnozowano MELAS | |
| Specjalność | neurologia |
genetyka
Biopsja mięśnia osoby, u której zdiagnozowano MELAS, ale bez znanej mutacji. (a) Zmodyfikowany trójkolorowy barwnik Gomori pokazuje trochę postrzępionych czerwonych włókien (strzałki). (b) Barwienie oksydazą cytochromu c, pokazujące włókna typu 1, lekko wybarwione i typu II, włókna ciemne i kilka włókien z nieprawidłowymi zbiorami mitochondrialnymi (strzałki). Należy zauważyć, że włókna ujemne oksydazy cytochromu c są zwykle obserwowane w encefalopatii mitochondrialnej, kwasicy mleczanowej i epizodach udaropodobnych (MELAS). (c) Barwienie dehydrogenazą bursztynianową pokazuje kilka postrzępionych niebieskich włókien i intensywne barwienie w mitochondriach naczyń krwionośnych (strzałka). (d) Mikroskopia elektronowa pokazuje nieprawidłowy zbiór mitochondriów z inkluzjami parakrystalicznymi (strzałki), inkluzjami osmiofilowymi (duże strzałki) i wakuolami mitochondrialnymi (małe strzałki).
MELAS jest spowodowany mutacjami w genach mitochondrialnego DNA.
dehydrogenazy NADH
Mutacje w MT-TL1 powodują ponad 80 procent wszystkich przypadków MELAS. Zmniejszają zdolność mitochondriów do wytwarzania białek, wykorzystywania tlenu i wytwarzania energii. Naukowcy nie ustalili, w jaki sposób zmiany w mitochondrialnym DNA prowadzą do określonych oznak i objawów MELAS. Nadal badają skutki mutacji genów mitochondrialnych w różnych tkankach, zwłaszcza w mózgu.
dziedzictwo
Ten stan jest dziedziczony według wzorca mitochondrialnego, który jest również znany jako dziedziczenie matczyne i heteroplazmia. Ten wzór dziedziczenia odnosi się do genów zawartych w mitochondrialnym DNA. Ponieważ komórki jajowe, ale nie plemniki, dostarczają mitochondriów do rozwijającego się zarodka, tylko samice przechodzą przez warunki mitochondrialne dla swoich dzieci. Zaburzenia mitochondrialne mogą pojawiać się w każdym pokoleniu rodziny i mogą dotyczyć zarówno mężczyzn, jak i kobiet, ale ojcowie nie przekazują mitochondrialnych cech swoich dzieci. W większości przypadków osoby z MELAS dziedziczą zmieniony gen mitochondrialny od matki. Rzadziej zaburzenie wynika z nowej mutacji w genie mitochondrialnym i występuje u osób bez rodzinnej historii MELAS.
diagnostyka
Leczenie / rokowanie
Pacjenci są leczeni w zależności od tego, jakie obszary ciała są dotknięte w danym momencie.