Melas sendromlu çocuklar ne kadar yaşar? Nadir hastalıklar. MELAS sendromunun belirtileri
anahtar kelimeler
MELAS SENDROMU / MELAS SENDROMU / EPİLEPSİ / EPİLEPSİ / KLİNİKdipnot klinik tıp üzerine bilimsel makale, bilimsel çalışmanın yazarı - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
MELAS sendromu, laktik asidoz ve felç benzeri ataklarla (mitokondriyal ensefalomyopati, inme benzeri ataklarla birlikte laktik asidoz) mitokondriyal ensefalomyopati olarak tanımlanan mitokondriyal hastalıklar grubundan genetik olarak belirlenmiş bir hastalıktır. Tüm organlar ve dokular patolojik sürece dahil olur, ancak kas ve sinir sistemleri daha fazla acı çeker. Hastalık en sık 6 ila 10 yaşları arasında gelişir. Hastalığın seyri ilerleyicidir. Çoğu durumda, hastalık epileptik nöbetler, tekrarlayan baş ağrıları, kusma ve iştahsızlık ile kendini gösterir. Epilepsi, MELAS sendromunun önemli bir klinik belirtisidir. Epileptik nöbetler mitokondriyal ensefalopatilerde (ME) vakaların %53'ünde tanınabilen ilk semptomdur. MELAS'ta oksipital epilepsi en yaygın olanıdır. Hastalığın ilerlemesiyle birlikte, epilepsinin tedaviye direnci, genellikle bir durum seyri ile not edilir. Kozhevnikov'un epilepsisine dönüşüm vakaları açıklanmaktadır. Yaşamı boyunca doğrulanmış MELAS sendromu tanısı olan bir hastanın vaka öyküsünü sunuyoruz.
İlgili konular klinik tıpta bilimsel makaleler, bilimsel çalışmanın yazarı - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
-
İnme benzeri ataklar ve laktik asidoz (melas sendromu) ile mitokondriyal ensefalopati: tanı kriterleri, epileptik nöbetlerin özellikleri ve klinik bir vaka örneğinde tedaviye yaklaşımlar
2017 / Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A. -
Mitokondriyal hastalıklarda felç
2012 / Pizova N.V. -
Mitokondriyal hastalıkları olan çocuklarda epilepsi: tanı ve tedavi özellikleri
2012 / Zavadenko N.N., Kholin A.A. -
Mitokondriyal ensefalomyopatide nörolojik bozukluklar - felç benzeri ataklarla (MELAS sendromu) laktik asidoz
2012 / Kharlamov Dmitry Alekseevich, Krapivkin Alexey Igorevich, Sukhorukov Vladimir Sergeevich, Kuftina Lyudmila Andreevna, Groznova Olga Sergeevna -
Alışılmadık bir hipoparatiroidizm nedeni olarak Melas sendromu: klinik bir vaka
2018 / Umyarova Dilyara Shamilevna, Grebennikova Tatyana Alekseevna, Zenkova Tatyana Stanislavovna, Sorkina Ekaterina Leonidovna, Belaya Zhanna Evgenievna -
Laktik asidozlu mitokondriyal ensefalomyopatide inme benzeri ataklar
2010 / Kalashnikova Lyudmila Andreevna, Dobrynina L.A., Sakharova A.V., Chaikovskaya R.P., Mir-kasimov M.F., Konovalov R.N., Shabalina A.A., Kostyreva M.V., Gnezditsky V.V., Protsky S.V. -
Mitokondriyal sitopatiler: melas ve MIDD sendromları. Tek bir genetik kusur, farklı klinik fenotipler
2017 / Muranova A.V., Strokov I.A. -
Erken başlangıçlı çocukluk çağının iyi huylu oksipital epilepsisi (Panayotopoulos sendromu). Klinik vakanın tanımı
2015 / Matyuk Yu.V., Kotov A.Ş., Borisova M.N., Panteleeva M.V., Shatalin A.V. -
POLG1 gen mutasyonu ile ilişkili ilerleyici mitokondriyal ensefalomyopatinin klinik belirtilerinin polimorfizmi
2016 / Yablonskaya M.I., Nikolaeva E.A., Shatalov P.A., Kharabadze M.N. -
Kalıtsal mitokondriyal hastalıklarda enzimlerin sitokimyasal aktivitesi çalışmasının tanı değeri
2017 / Kazantseva I.A., Kotov S.V., Borodataya E.V., Sidorova O.P., Kotov A.S.
MELAS SENDROMUNDA EPİLEPSİ
MELAS sendromu, mitokondriyal ensefalomyopati, felç benzeri ataklarla seyreden laktik asidoz olarak tanımlanan mitokondriyal grubun genetik olarak belirlenmiş bir hastalığıdır. Patolojik süreç tüm organları ve dokuları içerir, ancak çoğunlukla kas ve sinir sistemleri için olumsuzdur. Hastalık en sık 6-10 yaş arası çocuklarda görülür. Klinik seyir ilerleyicidir. Çoğu durumda hastalık, epileptik nöbetler, tekrarlayan baş ağrıları, kusma, anoreksi ile kendini gösterir. MELAS sendromunun önemli klinik prezentasyonu epilepsidir. Epileptik nöbetler, vakaların %53'ünde mitokondriyal ensefalopatilerin (ME) ilk tanı semptomudur. MELAS sendromunda en sık oksipital epilepsi görülür. Hastalık ilerledikçe, genellikle status epileptikusun ortaya çıkmasıyla birlikte epilepsinin tedaviye direnci gözlenir. Kozhevnikov epilepsisine dönüşen bazı vakalar anlatılmıştır.Yaşarken doğrulanmış bir MELAS sendromu tanısı olan bir hastanın öyküsü verilmiştir.
Bilimsel çalışmanın metni "Melas sendromunda epilepsi" konulu
CİLT IV SAYI 3 2009
MELAS SENDROMLU EPİLEPSİ
K.Yu. Muhin1, M.B. Mironov1, N.V. Nikiforova1, C.B. Mihaylova2, VA. Chadaev1, AA. Alikhanov1-2, B.N. Ryzhkov1, A.Ş. Petrukhin1
MELAS SENDROMUNDA EPİLEPSİ
KYu. Muhin1, M.B. Mironov1, N.V. Nikiforova1, S.V. Mihaylova2, Birleşik Arap Emirlikleri. Chadaev1, AA. Alikhanov1-2, B.N. Ryzkov1 AS. Petrukhin1
1 - Nöroloji ve Nöroşirürji Anabilim Dalı, Pediatri Fakültesi, Devlet Yüksek Mesleki Eğitim Eğitim Kurumu, Roszdrav Rus Devlet Tıp Üniversitesi
2 - Rus Çocuk Klinik Hastanesi
MELAS sendromu, laktik asidoz ve felç benzeri ataklarla (mitokondriyal ensefalomyopati, inme benzeri ataklarla birlikte laktik asit) mitokondriyal ensefalomyopati olarak tanımlanan mitokondriyal hastalıklar grubundan genetik olarak belirlenmiş bir hastalıktır. Tüm organlar ve dokular patolojik sürece dahil olur, ancak kas ve sinir sistemleri daha fazla acı çeker. Hastalık en sık 6 ila 10 yaşları arasında gelişir. Hastalığın seyri ilerleyicidir. Çoğu durumda, hastalık epileptik nöbetler, tekrarlayan baş ağrıları, kusma ve iştahsızlık ile kendini gösterir. Epilepsi, MELA sendromunun önemli bir klinik belirtisidir. Epileptik nöbetler mitokondriyal ensefalopatilerde (ME) vakaların %53'ünde tanınabilen ilk semptomdur. MELAS'ta oksipital epilepsi en yaygın olanıdır. Hastalığın ilerlemesiyle birlikte, epilepsinin tedaviye direnci, genellikle bir durum seyri ile not edilir. Kozhevnikov'un epilepsisine dönüşüm vakaları açıklanmaktadır. Yaşamı boyunca doğrulanmış MELAS sendromu tanısı olan bir hastanın vaka öyküsünü sunuyoruz.
Anahtar kelimeler: MELAS sendromu, epilepsi, klinik, tanı, tedavi.
MELAS sendromu, mitokondriyal ensefalomyopati, felç benzeri ataklarla seyreden laktik asidoz olarak tanımlanan mitokondriyal grubun genetik olarak belirlenmiş bir hastalığıdır. Patolojik süreç tüm organları ve dokuları içerir, ancak çoğunlukla kas ve sinir sistemleri için olumsuzdur. Hastalık en sık 6-10 yaş arası çocuklarda görülür. Klinik seyir ilerleyicidir. Çoğu durumda hastalık, epileptik nöbetler, tekrarlayan baş ağrıları, kusma, anoreksi ile kendini gösterir. MELAS sendromunun önemli klinik prezentasyonu epilepsidir. Epileptik nöbetler, vakaların %53'ünde mitokondriyal ensefalopatilerin (ME) ilk tanı semptomudur. MELAS sendromunda en sık oksipital epilepsi görülür. Hastalık ilerledikçe, genellikle status epileptikusun ortaya çıkmasıyla birlikte epilepsinin tedaviye direnci gözlenir. Kozhevnikov epilepsisine dönüşen bazı vakalar anlatılmıştır.Yaşarken doğrulanmış bir MELAS sendromu tanısı olan bir hastanın öyküsü verilmiştir.
Anahtar kelimeler: MELAS sendromu, epilepsi, klinik tablo, tanı, tedavi.
MELAS sendromu, laktik asidoz ve felç benzeri ataklarla (mitokondriyal ensefalomyopati, inme benzeri ataklarla birlikte laktik asidoz) mitokondriyal ensefalomyopati olarak tanımlanan mitokondriyal hastalıklar grubundan genetik olarak belirlenmiş bir hastalıktır.
MELAS sendromu ilk olarak S. Pavlakis ve arkadaşları tarafından bağımsız bir nozolojik form olarak tanımlandı. 1984 yılında. Bununla birlikte, bazı yazarlar, hastalığın daha önce "ailesel poliodistrofi, mitokondriyal miyopati, laktik asidemi" adı altında tanımlandığını öne sürmektedir.
Popülasyondaki prevalansı belirlenmemiştir. 2000 yılına kadar, yerli basın da dahil olmak üzere 120'den fazla MELAS sendromu gözlemi yayınlandı.
MELAS sendromu vakaların %25'inde yüksek riskle anneden kalıtılır, ancak hastaların %56-75'inde aile öyküsü yüklenmez. Hastalık, solunum zinciri komplekslerinin alt birimlerini kodlayan mitokondriyal DNA genlerindeki ve taşıma RNA genlerindeki (MT-ND1, MT-ND5, MT-TH, MT-TL1 ve MT-TV) mutasyonlarla ilişkilidir. MELAS sendromu vakalarının %80-90'ında hastalık, lösin transfer RNA'sını kodlayan MT-TL1 genindeki bir nokta mutasyonuna dayanmaktadır. Bu mutasyonla, adenin nükleotidi, 3243 pozisyonunda (A3243G) guanin ile değiştirilir ve bu, mitokondrideki tüm proteinlerin sentezini bozar.
Tüm organlar ve dokular patolojik sürece dahil olur, ancak kas ve sinir sistemleri daha fazla acı çeker.
Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova C.V., Chadaev V.A., Alikhanov A.A., Ryzhkov BN., Petrukhin A.S.
MELAS Sendromunda Rus Epilepsisi. zhur. det. Neur.: cilt IV, no. 3, 2009.
ORİJİNAL MAKALELER
konular en uçucu olarak. Klinik belirtilerin şiddeti, eşik etkisine (yaş, dokuların enerji ihtiyaçları), nükleer genlerin solunum zinciri sentezi üzerindeki kontrolüne, heteroplazmiye (dokulardaki farklı mutant mtDNA molekülleri içeriği) bağlıdır. MELAS sendromlu hastalarda çeşitli dokularda mutant mtDNA içeriğinin %93-96 olduğu gösterilmiştir. Proband aile üyelerinde, dokularda mutant mtDNA da tespit edilir, ancak içeriği önemli ölçüde daha düşüktür: hastalığın silinmiş formunda% 62-89, sendromun klinik belirtilerinin yokluğunda% 28'den 89'a.
Hastalık en sık 6 ila 10 yaşlarında gelişir, ancak daha erken (iki yıla kadar) veya daha sonra ilk kez - 21 ila 40 yıl arasında vakalar vardır. Hastalığın başlangıcından önce hastaların %90-100'ü normal olarak gelişir. Hastalığın seyri ilerleyicidir, erken başlangıçlı daha kötü huyludur.
Çoğu durumda, hastalık epileptik nöbetler, tekrarlayan baş ağrıları, kusma ve iştahsızlık ile kendini gösterir. Ayrıca sağlığın bozulması ve kas zayıflığının ortaya çıkması şeklinde fiziksel aktiviteye karşı hoşgörüsüzlüğe de dikkat etmelisiniz. Miyopatik semptom kompleksi, egzersiz intoleransı, kas zayıflığı, yorgunluk ve bazen kas hipotrofisi ile kendini gösterir.
Hastalık ilerledikçe, genellikle bunama gelişir. Serebellar ataksi, nörosensoriyel sağırlık ve periferik polinöropati gibi semptomlar daha az yaygındır.
İnme benzeri ataklar, tekrarlayan baş ağrısı atakları, baş dönmesi, fokal nörolojik semptomların gelişimi (parezi, hemianopsi) ve koma ile kendini gösterebilen karakteristiktir. Bu akut ataklar genellikle ateş veya araya giren enfeksiyonlar tarafından tetiklenir. Bu tezahürler oldukça hızlı bir gerileme (birkaç saatten birkaç haftaya kadar) ve ayrıca tekrarlama eğilimine sahip olabilir.
Epilepsi, genellikle MELAS'ın erken evrelerinde ortaya çıkan önemli bir klinik bulgudur. BT
özellikle atipik mitokondriyal ensefalopatide (ME) sıklıkla en belirgin nörolojik bulgudur. Epileptik nöbetler mitokondriyal ensefalopatilerde (ME) vakaların %53'ünde tanınabilen ilk semptomdur.
MELAS'ta oksipital epilepsi (SE) en yaygın olanıdır. Oksipital loblardan kaynaklanan fokal nöbetlerle karakterizedir. Nöbetler genellikle görme alanı kaybı gibi geçici veya kalıcı nörolojik semptomlarla ilişkilidir.
Oksipital korteksten kaynaklanan nöbetler, tezahürlerine göre öznel duyumlara (aura) ve kural olarak bir motor bileşeniyle klinik olarak saptanabilir semptomlara bölünür. Oksipital lobdan yayılan epileptik auralar arasında basit ve karmaşık görsel halüsinasyonlar, amaurosis bulunur. SE'ye özgü en tipik nöbetler, pozitif (parlamalar, ışık noktaları) ve negatif semptomlar (skotom, hemianopsi) olarak ortaya çıkabilen basit görsel halüsinasyonlardır. Çoğu zaman, görsel halüsinasyonlar, sabit veya yanıp sönen bir nokta veya ışık noktaları olarak tanımlanır. Kural olarak, nokta yeşilimsi bir renk tonu ile beyazdır. Ayrıca halüsinasyonlar çok renkli veya tek renkli olabilir. Halüsinasyonlar genellikle oksipital kortekste uyarılma odağının kontralateralindeki görsel alanlarda ortaya çıkar ve ardından yayılır. Ancak hastaların şikayetlerinde görsel auranın sıklıkla tespit edilmediği unutulmamalıdır.
Epileptik uyarım oksipito-temporal veya oksipito-parietal bölgelere yayıldığında karmaşık görsel halüsinasyonlar not edilir. Karmaşık görsel halüsinasyonlar insanlar, hayvan nesneleri veya sahneler şeklinde görünebilir, tanıdık veya yabancı, hoş veya korkutucu, korkutucu, basit veya grotesk olabilir, statik olabilir veya yatay bir düzlemde hareket edebilir ve kaybolabilir. Kural olarak, bir motor saldırının gelişmesinden önceki bir terminal semptomdur; ilk iktal semptom olabilir, ancak daha sık
CİLT IV SAYI 3 2009
temel halüsinasyonlar.
İktal amavroz, oksipital korteksten kaynaklanan özel, teşhis edilmesi son derece zor bir nöbet türüdür. Birçok yazara göre, bu, görsel halüsinasyonların yanı sıra oksipital lobun tahrişinin aynı sık görülen semptomudur, ancak çoğu zaman tanınmadan kalır. Genellikle hastalar bu semptomu atak yapısında ayrı ayrı ayırt etmezler. Görme kaybı, yanal alanların kaybı ile iki taraflı olarak ortaya çıkar. Saldırı odağının kontralateralinde olası homonim hemianopi. Hastaların duyumları onlar tarafından gözlerde kararma, "beyaz karanlık", renk algısının bozulması olarak tanımlanır. Belki de sözde status epilepticus amauroticus'un oluşumu ile bir statü kursu.
Oksipital nöbetler otonomik semptomlarla ortaya çıkabilir. Bunlar migren baş ağrısı, baş dönmesi, mide bulantısı ve kusmayı içerir. Yaygın bir semptom, atak sonrası migren benzeri baş ağrısıdır.
Oksipital kortekste sınırlı olarak meydana gelen nöbetlerin klinik belirtileri, gözlerin yana doğru kayması ile karakterizedir. Gözlerin sapması, başın yana sapması ile birlikte not edilebilir. Çoğu durumda, gözlerin kontralateral odağa doğru sapması not edilir. Ancak, gözlerin odaklanmaya doğru abdüksiyonu gözlendiğinde vakalar açıklanır. Ayrıca, "oksipital" nöbetlerin özelliklerinden biri, deşarjın beynin ön kısımlarına anlık dağılımıdır, klinik tablo ise kural olarak, belirgin bir motor bileşeni tarafından yönetilir. Olası tonik, tonik-klonik (hemikonvülsif ve ikincil jeneralize), otomotor nöbetler. Bu bağlamda, ilk klinik semptomları belirlemek önemlidir - bakışların motive olmayan ve ani durması, var olmayan nesnelere bakmak, mantıksız bir gülümseme, bitkisel belirtiler ve VEM yöntemini kullanarak birincil iktojenik bölgeyi mutlaka belgelemek.
Hastalığın ilerlemesiyle birlikte, epilepsinin tedaviye direnci, genellikle bir durum seyri ile not edilir. Kozhevnikov epilepsisine dönüşüm vakaları açıklanmaktadır. Bir dizi oto-
Rov, daha önce epileptik nöbet öyküsü olmayan MELAS'lı hastalarda ilk semptom olarak status epileptikus olasılığını açıklar. Ribacoba R. et al. yayınlarında, öncesinde migren baş ağrısı atakları olan fokal motor nöbetleri olan 4 epilepsi parsiyel continua gelişimi vakasını tanımlamaktadır. Miyazaki M. et al. MELAS'lı hastalarda epilepsi parsiyelis continua içinde devam eden fokal miyoklonus olasılığını gösterdi. Araki T. et al. 37 yaşında bir hastayı, bilinç dalgalanmaları şeklinde fokal nöbetlerin epileptik durumu, yana paroksismal göz deviasyonu atakları ile birlikte homonim hemianopsi ile gözlemledi. EEG, oksipital bölgede lokalize nöbetlerin devam eden EEG paternlerini kaydetti. MELAS'lı erişkin hastalarda, fokal motor nöbetlerin baskınlığı vardır, ancak EEG, oksipital bölgelerde çok bölgeli epileptiform aktivitenin baskın olduğunu gösterir.
Epileptiform aktivite, nöbetlerin başlamasından sonra vakaların %71'inde kaydedilir. MELAS sendromlu hastaların elektroensefalografik bir çalışması, oksipital bölgelerde epileptiform aktivite ile karakterize edilir. Bazı yazarlar bölgesel epileptiform bozuklukların görünümünü felçlerle ilişkilendirir. Fujimoto S.'nin çalışmasına göre, akut dönemde (yani, felç benzeri bir epizoddan sonraki 5 gün içinde), MELAS sendromlu incelenen hastaların çoğunda polipiklerle birlikte bölgesel yüksek genlikli delta dalgaları vardı. Yazarlar bu paterni inme benzeri epizodlar için patognomonik olarak değerlendirmeyi önermektedir. Oksipital bölgelere ek olarak epileptiform aktivite temporal bölgelere, bifrontal ve bilateral olarak arka bölgelere yayılabilir. Belki de ritmik fotostimülasyon sırasında bir fotoparoksismal yanıtın ortaya çıkması.
Önde gelen laboratuvar işareti, kandaki laktat seviyesindeki bir artıştır.
ORİJİNAL MAKALELER
laktik asidoz gelişimine yol açan 2.0 mmol / l'nin üzerinde wi.
Epilepsi meydana gelse bile, hastalığın erken evrelerinde beynin MRG'si önemsiz olabilir. Nörogörüntüleme yöntemleri serebral hemisferlerde (%80), daha az sıklıkla serebellum ve bazal ganglionlarda enfarktüs bölgelerini ortaya çıkarır. Bazal ganglionların kalsifikasyonu, serebral korteksin atrofisi de olabilir. Bir foton emisyon çalışmasında, beyin bilgisayarlı tomogramında enfarktüs bölgesinin (izotop sinyalindeki azalma) ortaya çıkmasından 3-16 gün önce izotop birikimi tespit edilir. Beynin MRG'si geçici olabilen baskın olarak oksipital loblarda bulunan lezyonları gösterir. Oksipital korteks ağırlıklı olarak etkilenir, beyaz cevher daha az hasar görür. T2 ağırlıklı görüntülerde, MELA'daki beyin lezyonları, sinyal yoğunluğunun arttığı alanlar olarak görünür. Geçici hiperintens alanlar, bazı yazarlar tarafından geri dönüşümlü vasküler ödem ile ilişkilendirilmiştir.
Anjiyografi genellikle vasküler anormallikleri ortaya çıkarmaz. Difüzyon ağırlıklı MRG, vazojenik ödem ile ilişkili değişiklikleri gösterir.
Histopatoloji: Kas biyopsisi, yırtık "kırmızı kenarlı" lifleri ortaya çıkarır. Beynin otopsisi, eski ve yeni enfarktüs odaklarının yanı sıra korteksin atrofisi ile fokal nekroz odaklarının bir kombinasyonu ile karakterize edilir.
Şu anda, terapi destekleyicidir. Tedavinin ana yönü, mitokondri ve solunum zincirinin enerji dengesini iyileştirmektir. Koenzim p10 (80-300 mg / gün), K1 ve KZ vitaminleri (25 mg / gün), süksinik asit (6 g / güne kadar), C vitamini (2-4 g / gün), riboflavin (100 mg / gün) uygulayın. gün) ve nikotinamid (1 g/gün'e kadar). Gelişmekte olan ikincil karnitin eksikliği ile bağlantılı olarak, hastalara L-karnitin (100 mg/kg/gün'e kadar) reçete edilir. Antioksidan tedavi olarak E vitamini (300-500 mg/gün) ve C vitamini (2-4 mg/gün) kullanılmaktadır.
MELA için genel kabul görmüş bir antiepileptik tedavi rejimi yoktur. Bazı yazarlar, enerji metabolizmasını engelleyebilecek ilaçları (barbitüratlar, valproik asit ilaçları; ayrıca diğer gruplardan bazı ilaçlar, örneğin kloramfenikol) hariç tutmayı önermektedir. Literatür, A3243C mutasyonlu MELA sendromunda valproik asit kullanımı ile birkaç izole nöbet şiddetlenmesi vakasını tanımlamaktadır. MELA sendromunda epilepsi tedavisinde ana AED'lerin ortalama terapötik dozlarda tegretol (veya trileptal), topamax, keppra olduğu kabul edilir. Doğru seçilmiş tedavi, ikincil jeneralize konvülsif nöbetlerin sıklığında önemli bir azalmaya yol açar. Ancak vejetatif-visseral ve görme fonksiyonlarında bozulma olan nöbetler genellikle tedaviye dirençlidir. Hastalığın son aşamasında epileptik nöbetlerin sıklığı azalabilir.
İşte yaşamı boyunca doğrulanmış MELAY sendromu tanısı olan bir hastanın vaka öyküsü.
11 yaşındaki hasta Ch.A., Pediatrik Nöroloji ve Epilepsi Merkezinde gözlemlendi. Başvuruda, yavaş yavaş konuşma becerileri kaybı, yürümeyi reddetme ile belirgin bir yürüyüş bozukluğu, görmede önemli bir azalma, kaprislilik ve olumsuz davranış şikayetleri yapıldı; yüz kaslarının seğirmesi, üst ve alt ekstremite kaslarının yanı sıra kısa süreli görme kaybı atakları şeklinde günlük seri saldırılar.
Hastalığın başlangıcı 5 yıl 9 aylıkken kaydedildi. İlk kez, tam sağlığın arka planına karşı, uykuya dalarken, şiddetli bir baş ağrısı ortaya çıktı, basit görsel halüsinasyonlar ("sarı ışın"), ardından gözlerin şiddetli bir dönüşü ve yana doğru ve genelleştirilmiş bir gelişme. tonik-klonik konvülsif nöbet, ardından kusma kaydedildi. 9 ay sonra aynı semptomlara sahip saldırılar tekrarladı ve hızlı bir şekilde seri bir karakter kazandı. Günde 400 mg dozda tegretol atanmasından sonra, atak sıklığı ayda 1 defaya düştü. Tegretol, 900 mg/gün dozunda Depakine Chrono ile değiştirildi ve buna karşı 6 ay boyunca klinik bir remisyon kaydedildi. Klinik semptom göz önüne alındığında
CİLT IV SAYI 3 2009
tomatik, nöbetlerin uykuya dalma dönemine hapsolması, hastanın zekasının normal olması, valproata pozitif reaksiyon göstermesi, idiyopatik oksipital epilepsi tanısı konuldu.
7 yaşında, ayda 1 kez aynı sıklıkta uykuya dalarken sekonder genelleme ile fokal versif nöbetler yeniden başladı. Depakine dozunun 1500 mg/gün'e çıkarılması nöbet sıklığında azalmaya yol açmadı. 75 mg/gün dozunda lamiktal eklendiğinde ataklar 4 ay durmuş, daha sonra aynı sıklıkta devam etmiştir. 8 yaşında, kısa süreli görme kaybı olan ataklar katıldı. 8 yıl 8 aydan itibaren uykuya dalmadan önce, atipik devamsızlıklar ortaya çıkmaya başladı: göz kapaklarının kapanmasıyla hızlı yanıp sönme ve göz kürelerinin kurumu yukarı; bilinç dalgalanır.
9 yaşında, birkaç gün süren, gözlerin önünde yanıp sönen bir "ışın" şeklinde basit görsel halüsinasyonlar, gözleri ve başı sağa çevirerek birden fazla seri atak ortaya çıktı. Uykuya dalmadan önce, bu tür saldırılar bazen yüzün azalmasıyla kendini gösteren fokal hemiklonik olanlara dönüştü.
sağda kas sistemi, başın sağa seğirmesi, sağ uzuvların klonları (koldan daha büyük). Bazen saldırıdan sonra şiddetli bir baş ağrısı ve kusma oldu. Aynı yaşta, engelleyici nöbetler ortaya çıktı: sağ ayağın baş parmağında tüyleri diken diken eden bir aura, ardından sağ bacağın kısa süreli zayıflığı ve sağ elin beceriksizliği. Topamax 100 mg/gün dozunda tedavi rejimine dahil edildi - 1 yıl boyunca epileptik nöbet olmadı.
Ayrıca, 9 yaşında, şiddetli baş ağrısı, kusma ve sağ taraflı hemiparezi gelişimi ile birlikte paroksismal koşullar ilk kez ortaya çıktı. Bazı durumlarda, bu tür koşullara birkaç dakikadan birkaç güne kadar süren amaurosis eşlik etti.
10.5 yaşında, başın sola çevrilmesi, göz kürelerinin sola sarsıntılı hareketleri, 5 s'ye kadar süren, günde 3 defaya kadar sıklık, uyku sırasında bile ataklar yeniden ortaya çıktı. Topamax dozu, önemli bir etki olmaksızın 150 mg/gün'e çıkarıldı. 10 yıl 10 ayda. şiddetli bir baş ağrısından sonra, dönüşümlü olarak
Pirinç. 1. Hasta Ch.A. 10 yıl. Teşhis: MEAE sendromu. Semptomatik fokal epilepsi.
Video-EEG izleme (2004): beynin ana aktivitesinde yaygın bir yavaşlamanın arka planına karşı, sol oksipital bölgede devam eden epileptiform aktivite kaydedildi. Bir atağın subklinik EEG paternleri de sol arka temporal bölgeye yayılarak sol oksipital bölgede kaydedilmiştir.
Pediatrik Nöroloji ve Epilepsi Merkezi
Profesör K.Yu'nun rehberliğinde. Mukhina, Aetei'de sinir sisteminin anksiyete bozukluklarının tanı ve tedavisi ile uğraşmaktadır, Aetian epilepsi formlarında uzmanlaşmıştır.
Ana yönler
faaliyetler:
Çocuklarda ve ergenlerde epilepsi
Baş ağrısı
Çocuklarda uyku bozuklukları
Tiki, enürezis
Çocukların yaşamın ilk ^ ayında muayenesi.
Merkezimizde yapılan muayeneler:
Çocuklarda sinir sistemi hastalıklarının tanı ve tedavisi
Epilepsinin tam teşhisi (ameliyat öncesi dahil) ve tedavisi
Nörolog ve epileptologların danışmanlığı
Bir çocuk doktoruna danışma (sık hasta çocuklar, gastroenteroloji vb.)
Bir psikiyatrist ve psikoloğun danışmanlığı.
Testlerle genetik danışma (karyotipleme dahil)
Video-EEG izleme (Merkezin özel donanımlı odalarında veya hastanın evini ziyaret ederek)
Bilgisayar (dijital) elektroensefalografi
Baş ve boyun damarlarının UZDG (ultrason dopplerografisi)
Ekoensefalografi (ECHO EG)
Sitemizde İnternet üzerinden "Rus Çocuk Nöroloji Dergisi" dergisine abone olabilirsiniz.
Merkezin çalışmaları ile ilgili detaylı bilgi 10:00 - 19:00 arası telefon ile:
Tel.: (+7495) 983-09-03; (+7926)290-50-30 Tel./Faks: (+7495) 394-82-52
Adres: st. Borisovskie Prudy, 13, bldg. 2. İnternet: www.epileptologist.ru E-posta: [e-posta korumalı](ayrıntılı bir rota haritası için web sitesine bakın)
CİLT IV SAYI 3 2009
seri hale gelen ve 48 saat süren fokal hemklonik ve sekonder jeneralize nöbetler. Frizium, topamax'a 10 mg/gün dozunda geçici bir pozitif etki ile eklendi.
8 yaşından itibaren, okul materyallerinin özümsenmesiyle ilgili zorluklar not edilmeye başlandı; azalmış hafıza. Artan yorgunluk, bitkinlik, zihinsel aktivitenin engellenmesi vardı. Çocuk kaprisli, sinirli, olumsuz oldu; ruh halinin arka planı azaldı. 9 yaşından itibaren bu semptomatolojide bir artış oldu.
Yaşam anamnezinden çocuğun ikinci normal gebelikten, ikinci dönem doğumdan doğduğu, doğum ağırlığı 2800 g, uzunluğu 53 cm olduğu biliniyor.Erken psikomotor ve konuşma gelişimi tamamen yaşına uygundu. Geçmiş hastalıklar: 6 yaşında su çiçeği, 6 yaşından itibaren sık akut solunum yolu viral enfeksiyonları (yılda 4 defaya kadar). Epilepsi ve diğer nörolojik hastalıklar için kalıtım yükü yoktur.
Muayene sırasında (11 yaşında), çocuğun durumu ağırdı; incelemeye olumsuz tepki verir. Bilinçli, yanlısı
uzay ve zaman. Son derece isteksiz bir şekilde iletişim kurar, talimatları izlemeyi reddeder. Sola spontan nistagmus, sağa dönüşle baş sol omuza eğik. Dil orta hattadır, faringeal refleks azalır; Disfaji ve dizartri not edilir. Görme azalır.
Orta derecede yaygın kas hipotonisi belirlenir. Tendon refleksleri eşit olarak azalır. Sağ uzuvlarda kas gücünde hafif bir azalma vardı. Patolojik ayak refleksleri tespit edilmedi. Duyarlılık ihlali için nesnel bir veri yoktur. Romberg testinde buna değmez. Yürümeyi reddediyor. Onu ayağa kaldırmaya çalıştığınızda ağlıyor, yere oturuyor. Parmak izi testi yaparken eksik. Yavaşça, tek kelimelerle, isteksizce konuşur.
Ek muayene yöntemleri. Video-EEG izleme (2004). Ana arka plan kayıt etkinliğinin önemli ölçüde yavaşlaması. Çalışma sırasında, sol oksipital bölgede, sol posterior temporal bölgeye yayılmış ve periyodik bir EEG paterni oluşumu ile devam eden epileptiform aktivite kaydedildi.
1993 doğumlu 16/12/05
Pirinç. 2. Hasta Ch.A. 11 yıl. Teşhis: MELAS sendromu. Semptomatik fokal epilepsi.
Video-EEG izleme, 1 yıl sonra (2005) dinamik olarak gerçekleştirildi: beynin arka plan aktivitesinde önemli bir yavaşlama. Uyku kaydı sırasında, sağ fronto-merkez bölgede tepe dalga aktivitesinin tespit edildiği yapıda, sağ fronto-merkez bölgede devam eden bir bölgesel yavaşlama kaydedilir.
ORİJİNAL MAKALELER
stupa (Şekil 1). Ayrıca, tek keskin dalgaların dahil edilmesiyle sağ fronto-merkez bölgede devam eden bölgesel yavaşlama belirlenir.
Dinamiklerde Video-EEG izleme (2005): Beynin arka plan aktivitesinde önemli yavaşlama. Çalışma, sağ fronto-merkez bölgede bölgesel yavaşlamaya devam ettiğini kaydetti. Sağ fronto-merkez bölgedeki bölgesel yavaşlama yapısında, tepe dalga aktivitesi ortaya çıkar (Şekil 2).
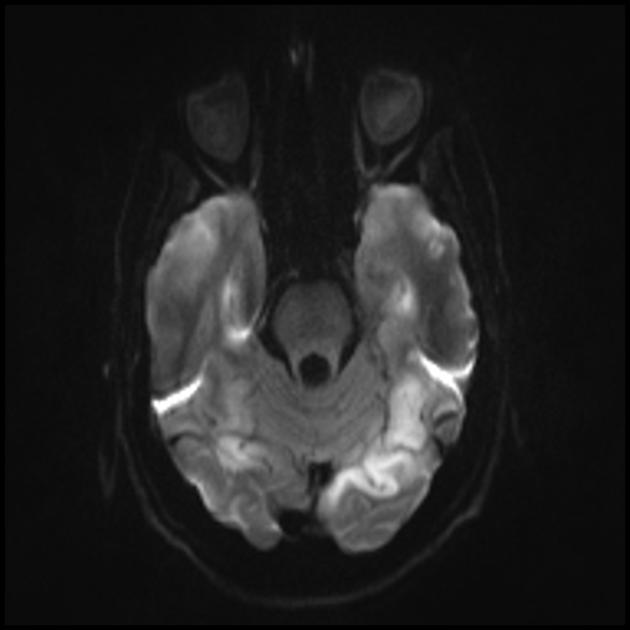
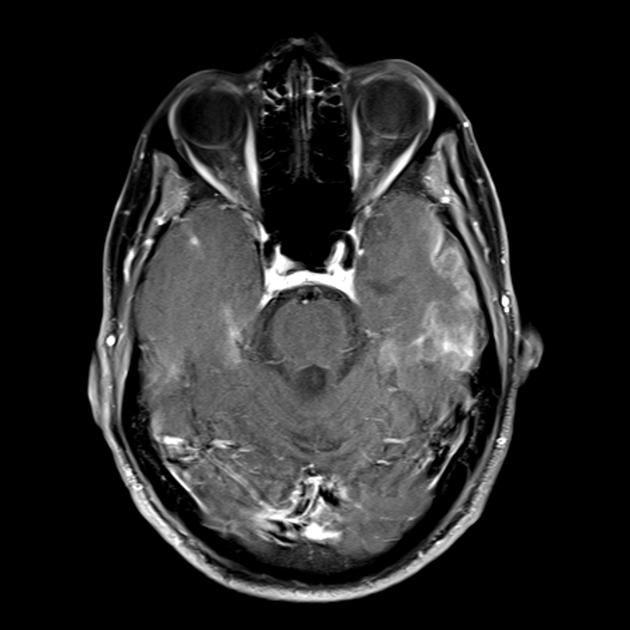
Beynin MRG'si. İlk MRG (6 yıl) serebellumun sol yarım küresinde T2 modunda tek bir hiperintens sinyali ortaya çıkardı. Zamanla MRG çalışması (10.5 yıl): patolojik sürecin beynin her iki yarım küresinin sol ve sağ oksipital-parietal bölgelerine yaygın olarak yayılmasıyla birincil lezyonda önemli bir bozulma ortaya çıktı (Profesör A.A. Alikhanov).
Görsel uyarılmış potansiyeller: optik sinir ve görsel analizörün kortikal kısmında görsel afferent sistemde önemli morfolojik ve fonksiyonel değişiklikler, solda daha belirgindir.
Göz doktorunun konsültasyonu: optik sinirlerin kısmi atrofisi. Kortikal agnozi unsurları.
Elektrokardiyogram: dakikada 100 vuruşa kadar hızlanma ile ektopik ritim.
Kalbin elektrik ekseninin dikey konumu. Ortostazda daha belirgin olan repolarizasyon süreçlerindeki değişiklikler.
Elektronöromyografi: lezyonun birincil kas tipini ortaya çıkardı. Periferik sinirler boyunca iletim hızları azalmaz.
Kandaki laktat seviyesinin incelenmesi: Kandaki laktat içeriği 3.0 mmol / l'dir (norm 1.8'e kadar).
Serebral korteksin oksipital bölgelerinden kaynaklanan, tedaviye dirençli, felç benzeri ataklar, amoroz dönemleri, bilişsel gerileme, beyincikte ve serebral korteksin arka bölgelerinde hiperintens sinyallerin varlığı dikkate alındığında, MRG'de kandaki laktat düzeyinde artış olan hastaya MELAS sendromu tanısı önerildi. Genetik muayene sırasında, kan hücrelerinde heteroplazmik durumda A3243G mutasyonu bulundu (tanı Rusya Tıp Bilimleri Akademisi Devlet Araştırma Merkezi'nde yapıldı) ve tanı doğrulandı.
Takipte gözlem, yüksek zihinsel işlevlerdeki ihlallerin hızlı bir şekilde ilerlediğini, kortikal körlüğün gelişimini, hastanın tamamen hareketsizliğini ve ardından 12 yaşında 10 aylıkken ölümün başladığını gösterdi. (hastalığın başlangıcından 7 yıl sonra).
bibliyografya
1. Nikolaeva E.A., Temin P.A. Bozulmuş nöropsikiyatrik gelişimin eşlik ettiği mitokondriyal hastalıklar. MELAS sendromu // Çocukların nöropsişik gelişiminin kalıtsal bozuklukları. Temin P.A. tarafından düzenlenen doktorlar için bir rehber. Kazantseva L.Z. - Tıp, 2001. - S. 96-107.
2. Nikolaeva E.A., Temin P.A., Nikanorova M.Yu., Klembovsky A.I., Sukhorukov V.S., Dorofeeva M.Yu., Korsunsky A.A. Mitokondriyal sendromlu bir çocuğun tedavisi MELAS (mitokondriyal ensefalopati, laktik asidoz, felç benzeri ataklar) // Rus Perinatoloji ve Pediatri Bülteni. - 1997. - No. 2. - S. 30-34.
3. Smirnova I.N., Kistenev B.A., Krotenkova M.V., Suslina ZA. Mitokondriyal ensefalomyopatinin (MELAS sendromu) felç benzeri seyri // Atmosfera. Sinir hastalıkları. - 2006. - Hayır. 1. - S. 43-48.
4. Temin PA, Nikanorova M.Yu., Nikolaeva E.A. MELAS sendromu (mitokondriyal ensefalomyopati, laktik asidoz, felç benzeri bölümler): ana belirtiler, tanı kriterleri, tedavi seçenekleri // Nevrol. dergi - 1998. - No. 2. - S. 43-48.
5. Ajmone-Marsan C., Ralston B. Epileptik nöbet, fonksiyonel morfolojisi ve tanısal önemi. - Springfield (IL): Charles C. Thomas, 1957. - S. 3-231.
6. Aldrich M.S., Vanderzant C.W., Alessi A.G., Abou-Khalil B., Sackellares J.C. Kalıcı görme kaybı olan iktal kortikal körlük // Epilepsi. - 1989. - C. 30. - S. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. Kompleks kısmi status epileptikus sunan bir MELAS vakası // Rinsho Shinkeigaku. - 2001. - V. 41(8). - S. 487-90.
CİLT IV SAYI 3 2009
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptic mitokondriyal bozukluklarla ilişkili fenotipler // Nöroloji. - 2001. - V. 56(10). - S. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. Valproik asit, A3243G mitokondriyal DNA mutasyonu olan bir hastada MELAS nedeniyle epilepsiyi şiddetlendirir // Metab Brain Dis. - 2007 - V. 22(1). - S.105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. ve diğerleri MELAS ve MERRF'nin moleküler patolojisi. Mutasyon yükü ile klinik fenotipler arasındaki ilişki // Beyin. - 1997. - V.120. - S. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Mitokondriyal DNA'nın (MELAS) A3243G mutasyonu ile ilişkili epilepsi polimorfizmi: gecikmiş tanı nedenleri // Rev Neurol (Paris). - 2004. - V. 160(8-9). - S. 824-829.
12. Dvorkin G., Andermann F., Carpenter S. Klasik migren, inatçı epilepsi ve çoklu felç: mitokondriyal ensefalopati ile ilgili bir sendrom / In: Andermann F., Lugaresi E., editörler. migren ve epilepsi. - Boston: Butterworths, 1987. - S. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Hastalarda seri elektroensefalograf bulguları MELAS // Pediatr Neurol ile. - 1999. - V. 20(1). - S. 43-48.
14. Goto Y., Nonaka I., Horai S.A. Mitokondriyal ensefalomiyopatilerin MELAS alt grubuyla ilişkili tRNA leu(UUR) genindeki bir mutasyon // Nature. - 1990. - V. 348. - S. 651-653.
15. Hasuo K., Tamura S., Yasumori K., Uchino A., Goda S., Ishimoto S., et al. MELAS'ta bilgisayarlı tomografi ve anjiyografi (mitokondriyal miyopati, ensefalopati, laktik asidoz ve felç benzeri ataklar): 3 vaka raporu // Nöroradyoloji. - 1987.-V. 29. - S. 393-397.
16. Hirano M., Pavlakis S.G. Mitokondriyal miyopati, ensefalopati, laktik asidoz ve felç benzeri ataklar (MELAS): Güncel kavramlar // J. clin. Nörol. - 1994. - V. 9. - S. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto, Hirose G. Mitokondriyal miyopati, ensefalopati, laktik asit ve felç benzeri atakları olan bir hastada epileptik nöbetler ( MELAS) // Jpn J Psikiyatri Nörol. - 1989. - V. 43(3). - S. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J., et al. Laktat-piruvat yükselmesi ve beyin enfarktüsü ile mitokondriyal ensefalomyopati // Nöroloji. - 1984. - V. 34. - S. 72-77.
19. Kuzniecky R. Semptomatik oksipital lob epilepsisi // Epilepsi. - 1998. - V. 39 Ek 4. - S. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Ekstratemporal kökenli nöbet bozukluklarında derinlik ve doğrudan kortikal kayıt // Nöroloji. - 1976. - V. 26. - S. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Oksipital elektroensefalografik odakları olan epileptik hastalarda klinik iktal paternler // Nöroloji. - 1975. - V. 25. - S. 463-471.
22. Matthews P.M., Tampieri D., Berkovic S.F., Andermann F., Silver K., Chityat D., et al. Manyetik rezonans görüntüleme, MELAS sendromunda // Nörolojide spesifik anormallikler gösterir. - 1991. - V. 41. - S. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. Epilepsia parsialis continua ile ilişkili MELAS'lı bir olgu // Hattatsu'ya Hayır. - 1991. - V. 23(1). - S.65-70.
24. Montagna P., Gallassi R., Medori R., Govoni E., Zeviani M., Di Mauro S., et al. MELAS sendromu: karakteristik migren ve epileptik özellikler ve anneden aktarım // Nöroloji. - 1988. - V. 38. - S. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N., et al. Mitokondriyal miyopati, ensefalopati, laktik asidoz ve felç benzeri bölümlerde serebral kan akışı // İnme. - 1993. - C. 24. - S. 304-309.
26. Pavlakis S.G., Phillips P.C., Di Mauro S. ve diğerleri. Mitokondriyal miyopati, ensefalopati, laktik asidoz ve felç benzeri bölümler: Ayırt edici bir klinik sendrom // Bir nörol. - 1984. - V. 16. - S. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. MELAS'ta status epilepticus'un özellikleri. Dört vakanın analizi // Nöroloji. - 2006. - V. 21(1). - S. 1-11.
28. Williamson P.D., Spencer S.S. Ekstratemporal kökenli karmaşık kısmi nöbetlerin klinik ve EEG özellikleri // Epilepsi. - 1986. - V. 27 (Ek 2). - S. 46-63.
29. Williamson P.D., Thadani V.M., Darcey T.M., Spencer D.D., Spencer S.S., Mattson R.H. Oksipital lob epilepsisi: klinik özellikler, nöbet yayılma paternleri ve cerrahi sonuçları // Ann Neurol. - 1992. - V. 31. - S. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Mitokondriyal DNA'nın A3243G mutasyonu olan bir hastada MELAS epilepsisini şiddetlendiren sodyum valproatın paradoksal etkisi // Central European Journal of Medicine. - 2007. - V. 2(1). - S.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. MELAS'ta vazojenik ödem: difüzyon ağırlıklı MR görüntüleme // Nöroloji ile seri bir çalışma. - 1999. - V. 53. - S. 2182-2184.
MELAS sendromu (MELAS) ilk olarak 1984 yılında S. Pavlakis ve arkadaşları tarafından tanımlanmıştır. Ancak bazı araştırmacılar, sendromun daha önce ailesel poliodistrofi, laktik asidemi gibi kavramlarla belirlenmiş olduğuna inanıyor.
patolojinin özü
1994 yılında, S. Pavlakis ve Mizio Hirano, hastalığın 110 vakasını tanımladı. MELAS (Mitokondriyal Ensefalomiyopati, Laktik Asidoz ve İnme benzeri ataklar) multisistem ilerleyici nörodejeneratif bir hastalıktır. Polimorfiktir ve konvülsiyonlar ve demans, laktik asidoz ile ensefalopati ile karakterizedir. Hastalığa mitokondriyal DNA'daki (mtDNA) mutasyonlar neden olur. Bu hastalığın başka bir adı var - mitokondriyal ensefalomyopati.
Genel bilgi
Hastalık vakalarının %25 ila %44'ü kalıtsaldır ve anne yoluyla bulaşır. Diğer durumlarda, ilk kez ortaya çıkar. Şu anda, mutasyona uğrayan ve bu sendromun gelişmesine yol açan 10'dan fazla gen bilinmektedir. Bunlar, transfer RNA'sının fonksiyonlarını kodlayan genlerdir. MELAS sendromu, anormal bir mitokondri birikimi ile hücrenin tüm enerji metabolizması sisteminin bozulmasına neden olan mitokondriyal hastalıkları (MD) ifade eder.
Bu grubun hastalıkları sadece anne yoluyla bulaşır. Onlarla birlikte, enerjiye en bağımlı organlar ve dokular farklı kombinasyonlarda etkilenir: iskelet kasları, kalp, beyin, görme, karaciğer ve böbrekler.
Semptomlar polimorfiktir ve her yaşta ortaya çıkabilir. Diyabet, işitme kaybı, nöbetler, endokrinopatiler, boy kısalığı, kardiyak patolojiler, mutlak egzersiz yapamama ve psikomotor anormalliklerin belirtilerini içerir.
Ondan önce psikomotor gelişim tamamen normaldir. Mutasyon, MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTTS2, MTND1, 5, 6. gibi birçok geni etkilediğinden, aynı semptomlara sahip hiçbir hasta tanımlanmadı. Sayıları artmaya devam ediyor.
Hastaların %80'inde MELAS sendromuna lösin tRNA genindeki (UUR) A3243G nokta ikamesi neden olur.
Frekans güvenilmez bir şekilde incelenmiştir. Sadece birkaç veri var: örneğin Finlandiya'da A3243G mutasyon oranı nüfusun 16:100 biniydi; İngiltere'de - 13 bin kişi başına 1 vaka.
patolojik değişiklikler

MELAS sendromunun karakteristik patolojik özelliği, özel bir Gomory üç renkli ile kas dokusunda görülebilen düzensiz kırmızı liflerdir (RRF). Bunlar, mutasyona uğramış genlerin ve bu anormal mitokondrilerin çoğalmasının bir sonucu olarak oluşan mtDNA hasarının morfolojik substratının sonucudur.
Zaten mitokondri nedir
Mitokondri, ana işlevi enerji sağlamak olan ökaryotik bir hücrenin (çekirdeği olan bir hücre) iki zarlı bir organelidir. Yani aslında mitokondri, hücrelerin enerji temeli, enerji istasyonlarıdır.
Hücrelerdeki mitokondri sayısı, yaşamı boyunca birkaç ile binlerce arasında değişebilir. Ve bunların çoğu, enerji üretimi ile ilişkili hücrelerde gerçekleşir.
Mitokondrilerin kendileri genellikle 1 ila 10 mikron arasında değişen, yuvarlak uzunlamasınadır. Hareketsizce donabilirler veya hücrenin sitoplazmasının içinde hareket edebilirler. Genellikle daha fazla enerjiye ihtiyaç duyulan yerlere giderler.
Mitokondrinin iç zarında, üzerinde tüm enzim sistemlerinin bulunduğu büyümeler (cristae) vardır. Temel olarak, bunlar protein bileşikleridir. Krista sayısı sentezleme işlemlerinin yoğunluğuna bağlıdır. Örneğin, kas hücrelerinin mitokondrilerinde her zaman birçoğu vardır.
Mitokondri, otonom bir protein sentez sistemine sahiptir - DNA, RNA ve ribozomlar. Gerekli proteinlerin bazıları mitokondri tarafından sentezlenir -% 5 ve bazıları sitoplazmadan elde edilir -% 95. Enerji, çeşitli enzimatik reaksiyonlar yoluyla organik bileşiklerden elde edilir.
Bu reaksiyonların bir kısmı oksijenin katılımı ile gerçekleşir, yani oksidasyon meydana gelir ve diğerlerinden sonra hidrojen protonlarının transferi ve enerjinin serbest bırakılması ile CO2 açığa çıkar. Başka bir deyişle, mitokondri, hücresel solunumda aktif bir katılımcıdır.
Bu reaksiyonlar cristae veya mitokondrinin kendisinde meydana gelir ki bu hücre için o kadar önemlidir ki iyileşirse hücre tamamen sağlıklı olur.
patogenez

İlk bakışta MELAS sendromunda durum inme sonrası bir varyantı andırır. Ancak aslında atipiktir: genellikle bulaşıcı hastalıklar tarafından kışkırtılan genç insanlarda görülür ve kötü huylu migren benzeri bir baş ağrısı, kasılmalar şeklinde ortaya çıkabilir.
Anjiyografi herhangi bir vasküler patoloji vermez. Normal damarlar olabilir veya bazı atardamarların, toplardamarların kalibrelerinde artış olabilir veya kılcal hiperemi meydana gelebilir.
MRI, MELAS sendromundaki akut beyin hasarının göç edebileceğini ve hatta ortadan kalkabileceğini göstermektedir. Bazı odaklar dalgalanıyor. Tipik bir felç için bu tamamen karakteristik değildir.
MELAS sendromunda multifokal nekroz varlığı vardır. Çoğunlukla, bu, serebral korteksin oksipital kısmında (arka lokalizasyon) ve subkorteksin beyaz maddesinde fark edilir. Ancak beynin diğer bölümlerinde de ortaya çıkabilirler. Bu alanlar kalp krizindeki nekroza benzer, ancak merkezi serebral damarların havzalarının dışında bulunur.
semptomatik belirtiler

Genellikle çocuklarda MELAS sendromu 6-10 yaşlarında başlar (3 yaşında ve 40 yaşında başlayabilir). Hastalığın erken başlangıcı daha tipiktir ve hastaların %90'ını etkiler. Erken bir başlangıçla, hastalık daha zor akar. Hastalar genellikle cılızdır, kasları zayıftır ve kesinlikle fiziksel efora adapte değildir.
Herhangi bir gerginlik veya fiziksel aktivite sizi daha kötü hissettirir. İç organlardan kalp, kas ve iletimin yetersiz beslenmesinden etkilenir, ardından kardiyovasküler yetmezlik gelişir. Nefropati, diyabet, kusma ile birlikte gastrointestinal rahatsızlıklar da meydana gelir, işitme azalır. Kas ağrısı, refleks eksikliği, parezi, konvülsiyonlar, IPE, bilinç kaybı ile karakterizedir. Kas zayıflığı (miyopatik sendrom) ve sensörinöral işitme kaybı da bu patoloji için tipiktir.
Endokrinopati sadece diabetes mellitus ile değil, aynı zamanda büyüme hormonu eksikliği ile de temsil edilir. Söz konusu hastalığın gelişiminde kalp ve böbrek rahatsızlıkları nadirdir.
MELAS sendromunda nöbetler oldukça değişkendir. Fokal, genelleştirilmiş, tonik-klonik ve miyoklonik olabilirler. Nöbetlerin antikonvülsan tedaviye mutlak duyarsızlığı karakteristiktir. Doktorların epilepsi teşhisi koyması ve örneğin valproik asit reçete etmesi sıklıkla olur. Ondan sonra, sağlık durumu keskin bir şekilde bozulur ve mitokondriyi baskıladığı için konvülsiyonlar artar. Demans gelişmesine rağmen, nadiren belirgin bir semptom haline gelir.
Aynı zamanda hastalığın özelliğidir, ancak diğer birçok patolojide de ortaya çıkar ve bu nedenle tanı için bir temel teşkil edemez. Sadece migren, konvülsiyon ve/veya felç benzeri fenomenlerle birleştiğinde MELAS sendromunun başlangıcından şüphelenilebilir. Bu kadar kapsamlı semptomlar bile doğru tanı koymaz. Sürecin ilerlemesi farklı şekillerde gerçekleşir.
işaretler

MELAS sendromunun ayırt edici klinik özelliği, nörolojik semptomların aniden ortaya çıktığı felç benzeri ataklardır (IPE). IPE, lezyonların asimetrisi ile karakterizedir. Çoklu olabilirler.
Bu tür bir lokalizasyonun seçiciliği ayrıca belirli odak semptomları verir:
- hemianopsi (kortikal körlük);
- hemiparezi;
- duyusal afazi (kelimelerin yanlış anlaşılması);
- acalculia (hesabın ihlali);
- agrafi (yazım ihlalleri);
- ataksi (gönüllü hareketlerin bozulmuş koordinasyonu);
- bilinç değişiklikleri.
Bu felç benzeri semptomların her 1 ila 3 ayda bir geri dönmesi nadir değildir. MELAS'taki akut atakların özelliği, hızlı bir gerileme olmaları, ancak çoğu zaman tekrarlamaları, yani iz bırakmadan geçmeleridir. Ek olarak, bu hastalığı olan hastalarda bazal ganglionlarda kalsifikasyonlar birikmektedir (bu, BT'de bulunur).
İnme benzeri ataklar genellikle 5-15 yaşlarında gelişir. Asla tromboembolizmin sonucu olmazlar. MELAS'taki anjiyopati, aynı mitokondrinin hiperproliferasyonundan kaynaklanır.
Semptomlarda IPE, tekrarlayan sefalji atakları, baş dönmesi, parezi, uzuvların felci, kraniyal sinirler ile kendini gösterir. Adam tamamen demoralize.
MELAS sendromunda laktik asidoz

Ana suçlusu, sinir sisteminin kanında ve dokularında aşırı miktarda laktik asittir. Bu, arterlerdeki kanın asitliğini keskin bir şekilde azaltır. Bu tür asidoz, MELAS sendromunda bulunan diabetes mellitusun sık görülen bir arkadaşıdır.
Erken bir aşamada, belirtiler spesifik değildir. Aşağıdaki belirtiler gözlenir: genel halsizlik, göğüs ağrısı, ilgisizlik, uyuşukluk. Fiziksel efordan sonra miyalji ve herhangi bir koku olmadan aralıklı hızlı nefes alma çok karakteristiktir.
Orta evrede laktik asit birikir ve hiperventilasyon sendromu (HVS) oluşur. Kanda karbondioksit birikir. Gürültülü solunum oluşmaya başlar - Kussmaul. Basınç çökecek kadar düşer, oligüri devreye girer. Hasta huzursuz, çılgın hale gelir ve daha sonra koma gelişmesiyle bilincini kaybeder - bu son aşamadır. Laktik asidoz belirtileri hızla, yani birkaç saat içinde gelişir. Sonra ölüm gelir.
Teşhis önlemleri

Daha önce belirtildiği gibi, semptomların polimorfizmi ve çok sayıda genin mutasyonu nedeniyle MELAS sendromunun teşhisi zordur. Kavradı:
- genel ve biyokimyasal kan testleri;
- kas biyopsisi;
- hasta akrabalar arasında karşılaştırmalı bir analiz ile genetik çalışma;
- Beynin BT'si: enfarktüs alanları daha sık hemisferlerde, daha az sıklıkla beyincikte, bazal ganglionlarda;
- kan damarlarının kalibresinde bir artış (arterler, damarlar, kılcal damarlar);
- DNA teşhisi: mtDNA'da karakteristik nokta mutasyonlarını arayın.
Terapi Yöntemleri
MELAS sendromunun tedavisi geliştirilmemiştir ve şu anda tedavi edilemez. Sadece yenilgi sürecini yavaşlatma girişimleri var. Tedavi iki yöne gider: sendrom sonrası tedavi (epilepsi, diabetes mellitus) ve patojenetik. Ancak günümüzde etkili bir patogenetik tedavi yoktur.
Semptomatik tedaviler vardır: işitme kaybı ile işitme cihazları aktif olarak kullanılır, solunum kas zayıflığı ile solunum tedavisi sağlanır. Hastanın kanındaki MELAS sendromu ile IPE sırasında L-arginin seviyesinin önemli ölçüde düştüğü kaydedildi. Bu nedenle tedavi arginin preparatları ve vitaminlerle yapılır. Koenzim Q veya idebinonun (noben), süksinik asit preparatlarının, K 1 ve K 3 vitaminlerinin, B 2 , B 3 , E, C'nin olumlu etkisi araştırılmaktadır; L-karnitin, antioksidanlar (meksidol, mildronat), laktik asidoz düzelticiler (dimefosfon) - hepsi hücrenin enerji metabolizmasını iyileştirir. Nöbetlerin tedavisinde, mitokondriyi baskıladıkları için valproatlar ve barbitüratlar reçete edilmez.
Bu sendromun önlenmesi için tüp bebek yöntemine başvurmak en iyisidir. Bir kadın ailesinde bu hastalığın tezahürü olduğunu biliyorsa, sağlıklı bir kadından döllenme için sitoplazma alınır. Yöntem henüz çalışma aşamasındadır, kütle değildir.
Bazı özellikler
Mitokondriyal bozuklukların teşhisi, tedaviye çok dikkatli bir yaklaşım gerektirir. Metabolik etki araçları mutlaka buna dahil edilmelidir. Hücrelerde doku solunumu, oksidatif fosforilasyon süreçlerini stabilize ederler. Sadece bu tür bir tedavinin sistematik olarak uygulanması, inme olaylarını önleyerek hastaların durumunun korunmasına yardımcı olabilir.
Tahmin etmek
Etkili tedavi eksikliği nedeniyle prognoz olumsuzdur. Semptomların ilk başlangıcından itibaren beklenen yaşam süresi genellikle beş yılı geçmez. Hastalığın nedenleri hakkında bilgi eksikliği, optimal tedavi rejiminin henüz bulunamamasına yol açmaktadır. Bütün bunlar tedavi şansını minimuma indirir.
MELAS sendromu, kas ve CNS tutulumu ile karakterize bir mitokondriyal bozukluktur.
MELAS (İng. Mitokondriyal ensefalomyopati, laktik asidoz ve felç benzeri ataklar - “mitokondriyal ensefalomyopati, laktik asidoz, felç benzeri ataklar”), başlıkta listelenen belirtilerle karakterize ilerleyici bir nörodejeneratif hastalıktır ve polimorfik semptomların eşlik ettiği - inme, diyabet, nöbetler, işitme kaybında azalma, kalp hastalığı, boy kısalığı, endokrinopatiler, egzersiz intoleransı ve nöropsikiyatrik bozukluklar.
Hikaye.
MELAS sendromu ilk olarak 1984 yılında Pavlakis ve arkadaşları tarafından tanımlanmıştır; on yıl sonra, Pavlakis ve Mizio Hirano 110 vakanın bir incelemesini yayınladılar.
miras türü:
anne
epidemiyoloji:
Hastalığın kesin sıklığı bilinmemektedir. Literatürde hastalığın insidansı ile ilgili çok az veri bulunmaktadır. Kuzey Finlandiya'da, A3243G mutasyon oranı 16.3:100.000'dir.
patogenez:
Mitokondrinin solunum zincirini kontrol eden mitokondral DNA mutasyonlarına, hücredeki metabolik süreçler için en önemli enerji kaynağı olan oksidatif fosforilasyon süreçlerinde bir bozulma eşlik eder.
Klinik bulgular
40 yaşında, MELAS'lı hastalar, epilepsi, tekrarlayan kusma, baş ağrısı ve kas güçsüzlüğünün yanı sıra geçici iskemik atak kliniği ile kabul edilir. Bu hastalara genellikle klinik olarak demans teşhisi konur.
Genç yaş ve inmeye özgü risk faktörlerinin olmaması MELAS'ı daha düşünceli kılmaktadır.
laboratuvar verileri
Laktat asidoz - artan laktat ve piruvat seviyeleri.
görselleştirme verileri
Beyindeki değişiklikler felçteki değişikliklere benzer.
Bir vuruştan farklılıklar
1) etkilenen alanlar arteriyel vasküler havuzların sınırları ile örtüşmez.
2) tekrarlanan saldırılarla odaklar farklı bir lokalizasyonda görselleştirilir.
+ klinik veriler (genç yaş, inme için risk faktörü yok).
BT
Vasküler yatakla tutarsız çoklu hipodens alanlar.
Bazal ganglionların kalsifikasyonu (en sık yaşlı hastalarda).
Atrofi, gerileme ve klinik iyileşmenin arka planında ortaya çıkar.
MR
akut enfarktüs
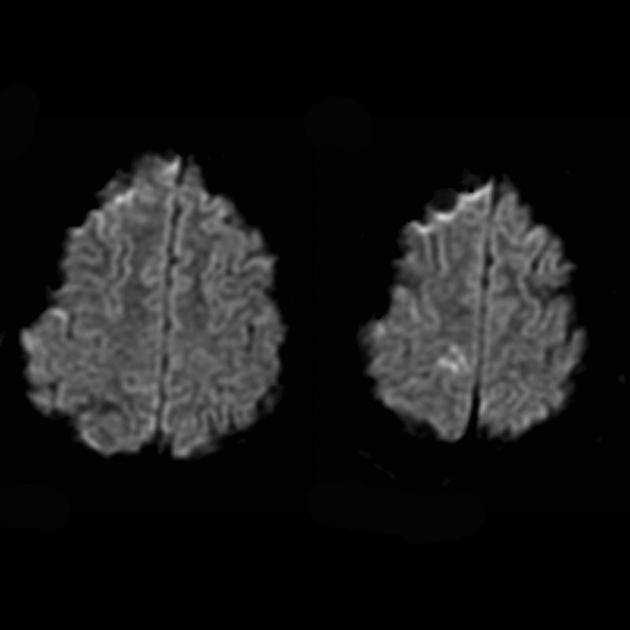
İnme ile ayrım için ADC ve DWI kullanılır (inmelerde difüzyon kısıtlaması (sitotoksik ödem) ve MELAS'ta difüzyon biraz sınırlıdır veya değişmez (vazojenik ödem).
Beynin subkortikal beyaz maddesinin patolojik sürecine katılım.
T2 ağırlıklı görüntülerde kıvrımların konturlarının netliğinin görselleştirilmesinde bozulma ve bunlardan gelen sinyalde artış.
kronik enfarktüs
Değişiklikler simetrik veya asimetrik olabilir.
Odak atrofisi, gerileme ve klinik iyileşmenin arka planında ortaya çıkar.
Beynin parietal, oksipital ve temporal lobları en sık etkilenir.
MR spektroskopisi
Artan laktat seviyeleri.




Materyaller nörologlar, terapistler ve genel pratisyenler için tasarlanmıştır.
Sergey Likhachev, baş, MD. bilimler, profesör;
Inessa Pleshko, Baş Araştırmacı, Ph.D. Bilimler, Cumhuriyet Bilimsel ve Pratik Nöroloji ve Nöroşirürji Merkezi Nöroloji Bölümü.
Subkortikal enfarktlar ve lökoensefalopati ile birlikte serebral otozomal dominant arteriyopati (CADASIL), klinik belirtileri tekrarlayan subkortikal iskemik inmeler, migren, subkortikal demans ve afektif bozuklukları içeren ilerleyici otozomal dominant bir hastalıktır. Mevcut yaygınlık - 1 vaka
100.000 nüfus başına.
Cumhuriyet Bilimsel ve Pratik Nöroloji ve Nöroşirürji Merkezi, CADASIL'li 7 hastayı (4 kadın dahil) görüyor; yaş - 32 ila 68 yaş arası. Nörolojik, moleküler genetik yöntemlerle incelendiler. Karakteristik semptomlar vardı; tarihte - migren, tekrarlayan laküner felçler ve afektif bozukluklar. Beyin MRG'si, CADASIL'in özelliği olan subkortikal enfarktları ve lökoensefalopatiyi ortaya çıkardı.
Moleküler genetik teşhis sonucunda 2 kişide 19. kromozomdaki Notch3 geninde CADASIL'e neden olan heterozigot mutasyon tespit edildi. Çentik genleri, hücre ontogenisi ile ilgili transmembran reseptörlerini kodlar. CADASIL ile çoğu durumda, transmembran proteinin yapısının değişmesi ve işlevlerinin bozulması nedeniyle yanlış anlamlı mutasyonlar belirlenir.
CADASIL'in patogenezi tam olarak açık değildir. Ana faktörün, beynin beyaz maddesinin küçük perforan damarlarının (kronik hipoperfüzyona yol açan) ilerleyici tıkanması ile arteriyopati olduğuna inanılmaktadır. Aynı zamanda, bazal membran bileşenlerinin çoğalmasına, orta zarın kalınlaşmasına ve küçük arterlerin mekanik sıkışmasına neden olan karakteristik granüler ozmiofilik kapanımlar bulunur. Sonuç olarak, kan-beyin bariyeri zarar görür - ödem gelişir.
Ek bir patolojik faktör, vasküler duvara yakın astrositlerin aktivasyonudur. Endotelyum-1'i serbest bırakarak vazokonstriksiyona ve kan akışının bozulmasına neden olurlar.
Granüler ozmiofilik inklüzyonların bileşimi bilinmemektedir. Notch3 proteininin bileşenlerinden biri olduğu varsayılmaktadır. Notch3 mutasyonu olan hastaların cilt biyopsilerinde, 20 yaşından önce bile ozmiofilik granüller ve düz kas hücrelerinin dejenerasyonu tespit edilebilir.
CADASIL'in klinik teşhisi:
- yüklü aile öyküsü;
- 50 yaşından önce hastalığın ilk belirtilerinin gelişmesi;
- aşağıdaki semptomlardan ikisinin varlığı - migren, tekrarlayan felçler, duygudurum bozuklukları, subkortikal demans.
Nörolojik semptomlarla etiyolojik olarak ilişkili vasküler risk faktörleri dışlanmalıdır. MRG, serebral hemisferlerin beyaz maddesindeki hasarı ve kortikal enfarktüslerin olmadığını gösterir.
Güvenilir bir "CADASIL" teşhisi, moleküler genetik teşhisin pozitif sonucu veya deri veya kas biyopsisinde karakteristik granüler ozmiofilik inklüzyonlar ile arteriyopatinin tespiti ile doğrulanır.
CADASIL'in en yaygın semptomları, hastaların neredeyse %85'inde gözlenen geçici iskemik ataklar ve iskemik inmelerdir.
Klasik laküner felç sendromları ve birkaç gün veya hafta sonra tam klinik remisyon ile kendini gösteren tekrarlayan bir seyir ile karakterize edilirler.
İkinci en yaygın olanı bilişsel bozukluklardır (hastaların %60'ında belirtilmiştir). 35 yaşında, hatta bazen iskemik ataklardan önce başlayabilir. CADASIL hastalarının yaklaşık %75'inde bunama gelişir. İlk semptom genellikle bir migrendir; genellikle 20 yaşından önce ortaya çıkar ve genellikle felçlerden önce gelir.
CADASIL'deki patolojik sürece kalbin katılımına ilişkin veriler çelişkilidir. L. Oberstein ve ark. (2003), CADASIL tanısı alan hastaların %25'inin elektrokardiyogramda akut miyokard enfarktüsü veya Q dalgası patolojisi öyküsü olduğunu saptamıştır. Başka bir çalışmada Cumurciuc ve ark. (2006), Notch3 mutasyonu olan 23 kişide pozitif kardiyak öykü bulamadı.
CADASIL'in klinik belirtileri ve farklı bir etiyolojiye sahip serebral mikroanjiyopati benzerdir - ayırıcı tanı gereklidir.
Hastalarda ve ailelerinde CADASIL'i zamanında belirlemek için moleküler genetik yöntemlere ve/veya histolojik çalışmalara başvurmak gerekir.
MELAS sendromu
Laktik asidoz ve felç benzeri ataklarla (MELAS) birlikte görülen mitokondriyal ensefalomyopati, mitokondriyal genomun patolojisi, bozulmuş enerji metabolizması ve enerjiye en bağımlı organ ve dokuların (CNS, kalp ve iskelet kasları, gözler, böbrekler, karaciğer, kemik iliği, endokrin sistem). MELAS sendromunun klinik belirtilerinin geniş değişkenliği ve nadir görülmesi, pratisyen için tanısal zorlukları önceden belirler.
Cumhuriyet Bilimsel ve Pratik Nöroloji ve Nöroşirürji Merkezi'nde 3 hastada (46 yaşında bir kadın ve 24 ve 23 yaşlarındaki oğulları) MELAS sendromu tanısı konuldu. Klinik ve nörolojik muayene, moleküler genetik teşhis, beyin MRG'sinden geçtiler.
Hepsi kısa; tarihte - mitokondriyal patolojinin belirtileri: sensörinöral işitme kaybı, migren benzeri baş ağrıları, zayıf egzersiz toleransı. Hastalığın başlangıcı genelleştirilmiş konvülsif nöbetlerdir. 2 hastada ilk belirtiler 20 yaşından önce ortaya çıktı; art arda epileptik nöbetler, oksipital ve temporal bölgelerde nörogörüntülemede odakların varlığı ile görme bozukluğu atakları, kan ve beyin omurilik sıvısında laktat düzeyinde artış vardı. 1 kişi bilişsel işlevlerde orta derecede azalma; kalbin ultrasonuna göre - hipertrofik kardiyomiyopati; diyabet.
Moleküler bir genetik çalışma, tRNA Leu(UUR) genindeki A3243G mutant kopyalarının sayısına karşılık gelen, MELAS için tipik olan çoklu sistem lezyonlarını, geniş değişkenliği ve değişen derecelerde klinik belirtileri ortaya çıkardı.
MELAS, anne tipi bir kalıtım, bir de novo mutasyon meydana geldiğinde sporadik vakaların varlığı ile karakterize edilir; hücrelerde - hem normal hem de mutant tiplerde - mitokondriyal DNA birikimi (heteroplazmi) ve yavru hücreler arasında bölünme sırasında rastgele dağılım (mitotik segregasyon). Genetik düzeyde, MELAS sendromunun nedeni, tRNALeu(UUR) genindeki 3243A>G heteroplasmik yeniden düzenlenmesidir (vakaların %80'i tespit edilir).
Hastalığın patogenezi henüz çalışılmamıştır. 2 ana teori vardır - "mitokondriyal anjiyopati" ve "mitokondriyal sitopati". İnme benzeri lezyonun vasküler zonlara tekabül etmediği ve uzamış epileptik aktivitenin neden olduğu vazojenik ödem eşlik etmesi nedeniyle çevre bölgelere yayıldığı bilinmektedir. Önerildiği gibi, felç benzeri bölümler, beynin sınırlı bir bölgesindeki sinirsel aşırı uyarılabilirlikten kaynaklanır. Kılcal endotel hücrelerinde veya nöronlarda veya astrositlerde mitokondriyal işlev bozukluğundan kaynaklanır; komşu nöronları depolarize ederek epileptik aktivitenin yayılmasına yol açar.
Ek olarak, tek foton emisyon bilgisayarlı tomografisine (SPECT) göre felç benzeri ataklar arasındaki aralıklarda, MELAS'lı hastalarda serebral hemodinamik bozukluğu gösteren posterior singulat korteks hipoperfüzyonu vardır.
Oksidatif fosforilasyonun ihlali, mitokondriyal solunum zincirinin yırtılması, katabolik metabolizmanın baskın olmasına ve Krebs döngüsünden laktat birikimi ile anaerobik glikoza değişikliklere katkıda bulunur. CNS'de yüksek düzeyde ikincisi genellikle nörolojik semptom dönemleri ile ilişkilidir.
MELAS'ın ana klinik belirtileri, felç benzeri ataklar, laktik asidoz ve kas biyopsi örneklerinde "yırtık kırmızı liflerin" varlığıdır. Ek belirtiler demans, psikoz, epileptik nöbetler, migren benzeri baş ağrıları, ataksi, miyopati, beyin görüntülemede bazal ganglionların kalsifikasyonu, optik atrofi, retinopati, sağırlık, diyabet, intestinal yalancı obstrüksiyon, kardiyomiyopati olabilir.
MELAS'ın ilk çıkışının erken yaşı 5 ila 20 yıldır, ancak geç başlangıçlı gözlemler vardır - yaşamın 5-6. on yıllarında. Sendromun kalp rahatsızlıklarından sonra başladığı durumlar vardır.
MELAS'taki lezyonların çoklu sistem yapısı klinik tanıyı zorlaştırır.
Hastalığın kalıtsal doğası, doğru tanı koymak için moleküler genetik çalışmalar yapmayı zorunlu kılmaktadır.
ve diğer hastaları tanımlayın - hastanın akrabaları arasından.
Materyaller nörologlar, terapistler ve genel pratisyenler için tasarlanmıştır.
| Mitokondriyal miyopati, ensefalomyopati, laktik asidoz ve felç benzeri ataklar | |
|---|---|
| Bazal ganglion kalsifikasyonu, serebellar atrofi, laktat artışı; MELAS teşhisi konan bir kişinin BT görüntüsü | |
| uzmanlık | nöroloji |
genetik
MELAS teşhisi konan ancak bilinen bir mutasyon taşımayan bir kişinin kas biyopsisi. (a) Gomori'nin modifiye edilmiş üç renkli boyasında bazı düzensiz kırmızı lifler (oklar) görülüyor. (b) Tip-1, hafif lekeli ve tip II lifleri, koyu lifleri ve anormal mitokondriyal koleksiyonları olan birkaç lifi gösteren sitokrom c oksidaz boyası (oklar). Sitokrom c oksidaz negatif liflerin genellikle mitokondriyal ensefalopati, laktik asidoz ve felç benzeri ataklarda (MELAS) görüldüğüne dikkat edin. (c) Süksinat dehidrojenaz boyaması, birkaç düzensiz mavi lif ve kan damarı mitokondrilerinde (ok) yoğun boyama gösterir. (d) Elektron mikroskopisi, parakristal inklüzyonlar (oklar), ozmiofilik inklüzyonlar (büyük oklar) ve mitokondriyal vakuoller (küçük oklar) ile anormal bir mitokondri koleksiyonunu gösterir.
MELAS, mitokondriyal DNA'daki genlerdeki mutasyonlardan kaynaklanır.
NADH dehidrojenazlar
mutasyonlar MT-TL1 tüm MELAS vakalarının yüzde 80'inden fazlasına neden olur. Mitokondrinin protein yapma, oksijen kullanma ve enerji üretme yeteneğini azaltırlar. Araştırmacılar, mitokondriyal DNA'daki değişikliklerin MELAS'ın spesifik belirti ve semptomlarına nasıl yol açtığını belirlemedi. Başta beyin olmak üzere çeşitli dokularda mitokondriyal gen mutasyonlarının etkilerini araştırmaya devam ediyorlar.
miras
Bu durum, aynı zamanda maternal kalıtım ve heteroplazmi olarak da bilinen mitokondriyal bir düzende kalıtılır. Bu kalıtım modeli, mitokondriyal DNA'da bulunan genleri ifade eder. Sperm değil de yumurtalar gelişen embriyoya mitokondriye katkıda bulunduğundan, sadece dişiler bebekleri için mitokondriyal koşullardan geçerler. Mitokondriyal bozukluklar bir ailenin her neslinde ortaya çıkabilir ve hem erkekleri hem de kadınları etkileyebilir, ancak babalar çocuklarının mitokondriyal özelliklerini aktarmaz. Çoğu durumda, MELAS'lı kişiler, değiştirilmiş mitokondriyal geni annelerinden miras alırlar. Daha az yaygın olarak, bozukluk mitokondriyal gendeki yeni bir mutasyondan kaynaklanır ve ailesinde MELAS öyküsü olmayan kişilerde görülür.
teşhis
Tedavi / prognoz
Hastalar, belirli bir zamanda vücudun hangi bölgelerinin etkilendiğine göre yönetilir.