Koliko dugo žive djeca sa melas sindromom? Rijetke bolesti. Simptomi MELAS sindroma
Ključne riječi
MELAS SINDROM / MELAS SINDROM / EPILEPSIJA / EPILEPSIJA / KLINIKAanotacija naučni članak o kliničkoj medicini, autor naučnog rada - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
MELAS sindrom je genetski uslovljena bolest iz grupe mitohondrijalnih bolesti, definisana kao mitohondrijalna encefalomiopatija sa laktacidozom i epizodama sličnim moždanom udaru (mitohondrijalna encefalomiopatija, laktacidoza sa epizodama sličnim moždanom udaru). U patološki proces su uključeni svi organi i tkiva, ali u većoj mjeri pate mišićni i nervni sistem. Bolest se najčešće razvija u dobi od 6 do 10 godina. Tok bolesti je progresivan. U većini slučajeva, bolest se manifestuje epileptičkim napadima, ponavljajućim glavoboljama, povraćanjem i anoreksijom. Epilepsija je važna klinička manifestacija MELAS sindroma. Epileptički napadi su prvi prepoznatljiv simptom kod mitohondrijalnih encefalopatija (ME) u 53% slučajeva. U MELAS-u, okcipitalna epilepsija je najčešća. Sa progresijom bolesti uočava se rezistencija epilepsije na terapiju, često sa statusnim tokom. Opisani su slučajevi transformacije u Koževnikovu epilepsiju. Prikazujemo istoriju bolesti pacijenta sa dijagnozom MELAS sindroma verifikovanom tokom života.
Povezane teme naučni radovi iz kliničke medicine, autor naučnog rada - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
-
Mitohondrijska encefalopatija s epizodama sličnim moždanom udaru i laktacidozom (melas sindrom): dijagnostički kriteriji, karakteristike epileptičkih napada i pristupi liječenju na primjeru kliničkog slučaja
2017 / Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A. -
Moždani udar u mitohondrijskim bolestima
2012 / Pizova N.V. -
Epilepsija kod djece s mitohondrijalnim bolestima: karakteristike dijagnoze i liječenja
2012 / Zavadenko N. N., Kholin A. A. -
Neurološki poremećaji u mitohondrijalnoj encefalomiopatiji - laktacidoza s epizodama sličnim moždanom udaru (MELAS sindrom)
2012 / Kharlamov Dmitrij Aleksejevič, Krapivkin Aleksej Igorevič, Suhorukov Vladimir Sergejevič, Kuftina Ljudmila Andrejevna, Groznova Olga Sergejevna -
Melasov sindrom kao neobičan uzrok hipoparatireoze: klinički slučaj
2018 / Umyarova Dilyara Shamilevna, Grebennikova Tatyana Alekseevna, Zenkova Tatyana Stanislavovna, Sorkina Ekaterina Leonidovna, Zhanna Belaya -
Epizode slične moždanom udaru u mitohondrijalnoj encefalomiopatiji sa laktacidozom
2010. / Kalašnjikova Ljudmila Andreevna, Dobrinina L. A., Saharova A. V., Chaikovskaya R. P., Mir-kasimov M. F., Konovalov R. N., Shabalina A. A., Kostyreva M. V., Gnezditskij V. V., Protsky V. -
Mitohondrijalne citopatije: melas i MIDD sindromi. Jedan genetski defekt, različiti klinički fenotipovi
2017 / Muranova A.V., Strokov I.A. -
Benigna okcipitalna epilepsija djetinjstva s ranim početkom (Panayotopoulosov sindrom). Opis kliničkog slučaja
2015 / Matyuk Yu.V., Kotov A.S., Borisova M.N., Panteleeva M.V., Shatalin A.V. -
Polimorfizam kliničkih manifestacija progresivne mitohondrijske encefalomiopatije povezane s mutacijom gena POLG1
2016 / Yablonskaya M.I., Nikolaeva E.A., Shatalov P.A., Kharabadze M.N. -
Dijagnostička vrijednost proučavanja citokemijske aktivnosti enzima u nasljednim mitohondrijskim bolestima
2017 / Kazantseva I.A., Kotov S.V., Borodataya E.V., Sidorova O.P., Kotov A.S.
EPILEPSIJA U MELASOM SINDROMU
MELAS sindrom je genetski uvjetovana bolest mitohondrijalne grupe, definirana kao mitohondrijska encefalomiopatija, laktacidoza s epizodama sličnim moždanom udaru. Patološki proces zahvata sve organe i tkiva, ali je uglavnom negativan za mišićni i nervni sistem. Bolest je najčešća kod djece uzrasta od 6 do 10 godina. Klinički tok je progresivan. U većini slučajeva bolest se manifestuje epileptičkim napadima, povratnim glavoboljama, povraćanjem, anoreksijom. Važna klinička slika MELAS sindroma je epilepsija. Epileptički napadi su početni dijagnostički simptom mitohondrijske encefalopatije (ME) u 53% slučajeva. Okcipitalna epilepsija je najčešća kod MELAS sindroma. Kako bolest napreduje, uočava se rezistencija epilepsije na liječenje, često uz pojavu epileptičnog statusa. Opisani su neki slučajevi transformacije u Koževnikovu epilepsiju.Data je anamneza pacijenta sa verifikovanom dok je živ dijagnozom MELAS sindroma.
Tekst naučnog rada na temu "Epilepsija u melas sindromu"
TOM IV BROJ 3 2009
EPILEPSIJA SA MELAS SINDROMOM
K.Yu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, C.B. Mihailova2, VA. Čadaev1, AA. Alikhanov1-2, B.N. Ryzhkov1, A.S. Petrukhin1
EPILEPSIJA U MELASOM SINDROMU
KYu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, S.V. Mikhailova2, UA. Čadaev1, AA. Alikhanov1-2, B.N. Ryzkov1 AS. Petrukhin1
1 - Katedra za neurologiju i neurohirurgiju, Pedijatrijski fakultet, Državna obrazovna ustanova visokog stručnog obrazovanja, Ruski državni medicinski univerzitet Roszdrav
2 - Ruska dječja klinička bolnica
MELAS sindrom je genetski uslovljena bolest iz grupe mitohondrijalnih bolesti koja se definiše kao mitohondrijalna encefalomiopatija sa laktacidozom i epizodama sličnim moždanom udaru (mitohondrijalna encefalomiopatija, mlečna kiselina sa epizodama sličnim moždanom udaru). U patološki proces su uključeni svi organi i tkiva, ali u većoj mjeri pate mišićni i nervni sistem. Bolest se najčešće razvija u dobi od 6 do 10 godina. Tok bolesti je progresivan. U većini slučajeva, bolest se manifestuje epileptičkim napadima, ponavljajućim glavoboljama, povraćanjem i anoreksijom. Epilepsija je važna klinička manifestacija MELA sindroma. Epileptički napadi su prvi prepoznatljiv simptom kod mitohondrijalnih encefalopatija (ME) u 53% slučajeva. U MELAS-u, okcipitalna epilepsija je najčešća. Sa progresijom bolesti uočava se rezistencija epilepsije na terapiju, često sa statusnim tokom. Opisani su slučajevi transformacije u Koževnikovu epilepsiju. Prikazujemo istoriju bolesti pacijenta sa dijagnozom MELAS sindroma verifikovanom tokom života.
Ključne riječi: MELAS sindrom, epilepsija, klinika, dijagnostika, liječenje.
MELAS sindrom je genetski uvjetovana bolest mitohondrijalne grupe, definirana kao mitohondrijska encefalomiopatija, laktacidoza s epizodama sličnim moždanom udaru. Patološki proces zahvata sve organe i tkiva, ali je uglavnom negativan za mišićni i nervni sistem. Bolest je najčešća kod djece uzrasta od 6 do 10 godina. Klinički tok je progresivan. U većini slučajeva bolest se manifestuje epileptičkim napadima, povratnim glavoboljama, povraćanjem, anoreksijom. Važna klinička slika MELAS sindroma je epilepsija. Epileptički napadi su početni dijagnostički simptom mitohondrijske encefalopatije (ME) u 53% slučajeva. Okcipitalna epilepsija je najčešća kod MELAS sindroma. Kako bolest napreduje, uočava se rezistencija epilepsije na liječenje, često uz pojavu epileptičnog statusa. Opisani su neki slučajevi transformacije u Koževnikovu epilepsiju.Data je anamneza pacijenta sa verifikovanom dok je živ dijagnozom MELAS sindroma.
Ključne riječi: MELAS sindrom, epilepsija, klinička slika, dijagnostika, liječenje.
MELAS sindrom je genetski uslovljena bolest iz grupe mitohondrijalnih bolesti, definisana kao mitohondrijalna encefalomiopatija sa laktacidozom i epizodama sličnim moždanom udaru (mitohondrijalna encefalomiopatija, laktacidoza sa epizodama sličnim moždanom udaru).
MELAS sindrom su prvi identifikovali kao nezavisni nosološki oblik od strane S. Pavlakis i sar. 1984. godine. Međutim, brojni autori sugeriraju da je bolest ranije opisana pod nazivom "porodična poliodistrofija, mitohondrijska miopatija, mliječna acidemija".
Prevalencija u populaciji nije utvrđena. Do 2000. godine objavljeno je više od 120 opažanja sindroma MELAS, uključujući i domaću štampu.
MELAS sindrom je u 25% slučajeva naslijeđen po majci sa visokim rizikom, ali kod 56-75% pacijenata porodična anamneza nije opterećena. Bolest je povezana s mutacijama u genima mitohondrijske DNK koji kodiraju podjedinice kompleksa respiratornog lanca i transportne RNK gena (MT-ND1, MT-ND5, MT-TH, MT-TL1 i MT-TV). U 80-90% slučajeva MELAS sindroma, bolest je zasnovana na tačkastoj mutaciji u MT-TL1 genu koji kodira RNA za prijenos leucina. Ovom mutacijom, adenin nukleotid je zamijenjen gvaninom na poziciji 3243 (A3243G), što remeti sintezu svih proteina u mitohondrijima.
U patološki proces su uključeni svi organi i tkiva, ali u većoj mjeri pate mišićni i nervni sistem.
Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova C.V., Chadaev V.A., Alikhanov A.A., Ryzhkov B.N., Petrukhin A.S.
Epilepsija u sindromu MELAS Rus. zhur. det. Neur.: tom IV, br. 3, 2009.
ORIGINALNI ČLANCI
teme kao najpromenljivije. Ozbiljnost kliničkih manifestacija zavisi od graničnog efekta (starost, energetske potrebe tkiva), od kontrole nuklearnih gena nad sintezom respiratornog lanca, heteroplazme (različiti sadržaj mutantnih mtDNA molekula u tkivima). Pokazalo se da kod pacijenata sa MELAS sindromom sadržaj mutantne mtDNK u različitim tkivima iznosi 93-96%. Kod članova porodice probanda, mutantna mtDNK se takođe otkriva u tkivima, ali je njen sadržaj znatno manji: 62-89% kod izbrisanog oblika bolesti, od 28 do 89% u odsustvu kliničkih znakova sindroma.
Bolest se najčešće razvija u dobi od 6 do 10 godina, ali postoje slučajevi ranijeg (do dvije godine) ili kasnijeg debija - od 21 do 40 godina. Prije početka bolesti, 90-100% pacijenata se normalno razvija. Tok bolesti je progresivan, maligniji sa ranim početkom.
U većini slučajeva, bolest se manifestuje epileptičkim napadima, ponavljajućim glavoboljama, povraćanjem i anoreksijom. Treba obratiti pažnju i na netoleranciju na fizičku aktivnost u vidu pogoršanja zdravlja i pojave mišićne slabosti. Kompleks miopatskih simptoma manifestuje se netolerancijom na fizičku aktivnost, slabošću mišića, umorom, a ponekad i hipotrofijom mišića.
Kako bolest napreduje, obično se razvija demencija. Simptomi kao što su cerebelarna ataksija, neurosenzorna gluvoća i periferna polineuropatija su rjeđi.
Karakteristične su epizode slične moždanom udaru, koje se mogu manifestirati ponavljajućim napadima glavobolje, vrtoglavice, razvojem fokalnih neuroloških simptoma (pareza, hemianopsija) i komom. Ove akutne epizode često su izazvane groznicom ili interkurentnim infekcijama. Ove manifestacije mogu imati prilično brzu regresiju (od nekoliko sati do nekoliko sedmica), kao i tendenciju recidiva.
Epilepsija je važna klinička manifestacija koja se često javlja u ranim fazama MELAS-a. Ovo je
često najočitija neurološka manifestacija, posebno kod atipične mitohondrijalne encefalopatije (ME). Epileptički napadi su prvi prepoznatljiv simptom kod mitohondrijalnih encefalopatija (ME) u 53% slučajeva.
Kod MELAS-a, okcipitalna epilepsija (SE) je najčešća. Karakteriziraju ga fokalni napadi koji potiču iz okcipitalnih režnjeva. Napadi su često povezani s prolaznim ili upornim neurološkim simptomima kao što je gubitak vidnog polja.
Napadi koji proizlaze iz okcipitalnog korteksa dijele se prema svojim manifestacijama na subjektivne senzacije (aura) i na klinički uočljive simptome, u pravilu, s motoričkom komponentom. Epileptičke aure koje izlaze iz okcipitalnog režnja uključuju jednostavne i složene vizualne halucinacije, amaurozu. Najtipičniji napadaji karakteristični za SE su jednostavne vizualne halucinacije, koje se mogu manifestirati kao pozitivni (bljeskovi, svjetlosne mrlje) i negativni simptomi (skotom, hemianopsija). Najčešće se vizuelne halucinacije opisuju kao pjega ili mrlje svjetlosti, bilo stalne ili bljeskajuće. Po pravilu, mrlja je bijela sa zelenkastom nijansom. Takođe, halucinacije mogu biti višebojne ili jednobojne. Halucinacije se obično pojavljuju u vidnim poljima kontralateralno od žarišta ekscitacije u okcipitalnom korteksu s naknadnim širenjem. Međutim, treba napomenuti da se u pritužbama pacijenata vizualna aura ne otkriva često.
Složene vizualne halucinacije se primjećuju kada se epileptička ekscitacija proširi na zatiljno-temporalne ili zatiljno-parijetalne regije. Složene vizualne halucinacije mogu se pojaviti u obliku ljudi, životinjskih predmeta ili prizora, biti poznate ili nepoznate, prijatne ili zastrašujuće, zastrašujuće, jednostavne ili groteskne, mogu biti statične ili se kretati u horizontalnoj ravni i nestati. U pravilu su terminalni simptom prije razvoja motoričkog napada; može biti prvi iktalni simptom, ali se češće javlja nakon toga
TOM IV BROJ 3 2009
osnovne halucinacije.
Iktalna ama vroza je poseban, izuzetno težak za dijagnosticiranje tip napadaja koji potiče iz okcipitalnog korteksa. Prema mnogim autorima, ovo je isti čest simptom iritacije okcipitalnog režnja, kao i vizualne halucinacije, ali često ostaje neprepoznat. Obično pacijenti ne razlikuju ovaj simptom zasebno u strukturi napada. Gubitak vida se javlja bilateralno sa gubitkom bočnih polja. Moguća homonimna hemianopija kontralateralna od žarišta napada. Senzacije pacijenata opisuju kao zamračenje u očima, "bijeli mrak", poremećenu percepciju boja. Možda statusni tok sa formiranjem takozvanog status epilepticus amauroticus.
Okcipitalni napadi mogu se manifestovati autonomnim simptomima. To uključuje migrensku glavobolju, vrtoglavicu, mučninu i povraćanje. Čest simptom je glavobolja nalik migreni nakon napada.
Kliničke manifestacije napadaja koji se ograničeno javljaju u okcipitalnom korteksu karakteriziraju odstupanje očiju u stranu. Može se uočiti devijacija očiju zajedno sa devijacijom glave u stranu. U većini slučajeva primjećuje se devijacija očiju prema kontralateralnom fokusu. Međutim, opisani su slučajevi kada se opaža otmica očiju prema fokusu. Također, jedna od karakteristika "okcipitalnih" napada je trenutna distribucija iscjetka na prednje dijelove mozga, dok kliničkom slikom, po pravilu, dominira izražena motorička komponenta. Mogući tonični, toničko-klonični (i hemikonvulzivni i sekundarno generalizirani), automotorni napadi. S tim u vezi, važno je identificirati početne kliničke simptome - nemotivisano i naglo zaustavljanje pogleda, gledanje u nepostojeće predmete, nerazuman osmijeh, vegetativne manifestacije i obavezno dokumentiranje primarne iktogene zone VEM metodom.
Sa progresijom bolesti uočava se rezistencija epilepsije na terapiju, često sa statusnim tokom. Opisani su slučajevi transformacije u Koževnikovu epilepsiju. Brojni auto-
Rov opisuje mogućnost epileptičnog statusa kao prvog simptoma kod pacijenata sa MELAS-om bez anamneze prethodnih epileptičkih napadaja. Ribacoba R. et al. opisuju u svojoj publikaciji 4 slučaja razvoja epilepsije partialis continua sa fokalnim motoričkim napadima, čemu su u anamnezi prethodile epizode migrenske glavobolje. Miyazaki M. et al. pokazala mogućnost kontinuiranog fokalnog mioklonusa unutar epilepsije partialis continua kod pacijenata sa MELAS-om. Araki T. et al. promatrana bolesnica u dobi od 37 godina sa epileptičkim statusom fokalnih napadaja u vidu fluktuacija svijesti, homonimne hemianopsije u kombinaciji s paroksizmalnim epizodama devijacije oka u stranu. EEG je zabilježio nastavak EEG obrazaca napadaja lokaliziranih u okcipitalnoj regiji. Kod odraslih pacijenata sa MELAS-om dominiraju fokalni motorni napadi, ali EEG pokazuje prevagu multiregionalne epileptiformne aktivnosti u okcipitalnim regijama.
Epileptiformna aktivnost se bilježi u 71% slučajeva nakon pojave napadaja. Elektroencefalografska studija pacijenata sa MELAS sindromom karakterizirana je epileptiformnom aktivnošću u okcipitalnim regijama. Brojni autori povezuju pojavu regionalnih epileptiformnih poremećaja sa moždanim udarom. Prema studiji Fujimoto S., u akutnom periodu (tj. u roku od 5 dana nakon epizode slične moždanom udaru) većina pregledanih pacijenata sa MELAS sindromom imala je regionalne delta talase visoke amplitude u kombinaciji sa polispikovima. Autori predlažu da se ovaj obrazac smatra patognomoničan za epizode slične moždanom udaru. Osim na okcipitalne regije, epileptiformna aktivnost se može širiti i na temporalne regije, bifrontalno, ali i bilateralno na stražnje regije sa difuznom distribucijom. Možda pojava fotoparoksizmalne reakcije tokom ritmičke fotostimulacije.
Vodeći laboratorijski znak je povećanje nivoa laktata u krvi.
ORIGINALNI ČLANCI
wi preko 2,0 mmol/l, što dovodi do razvoja laktacidoze.
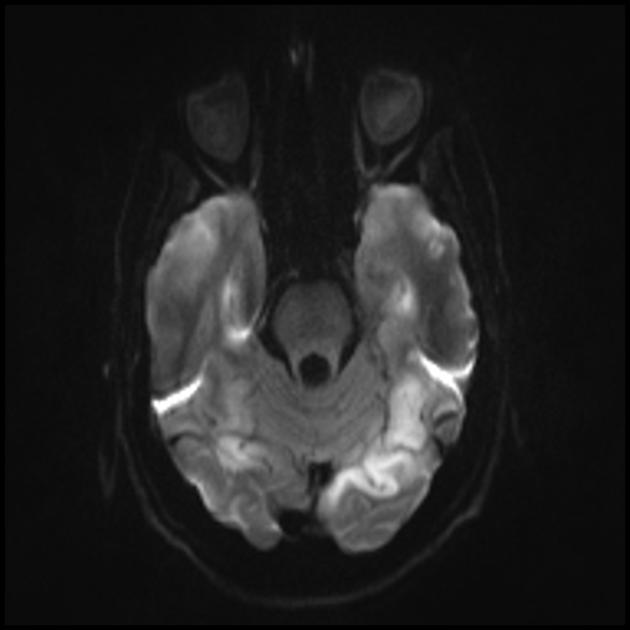
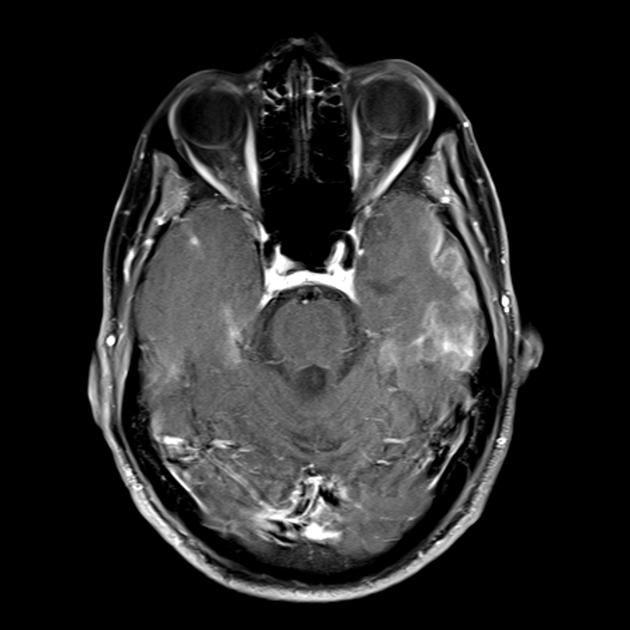
MRI mozga u ranim stadijumima bolesti može biti beznačajan, čak i ako se pojavi epilepsija. Neuroimaging metode otkrivaju infarktne zone u hemisferama mozga (80%), rjeđe u malom mozgu i bazalnim ganglijima. Može doći i do kalcifikacije bazalnih ganglija, atrofije moždane kore. U studiji emisije fotona, akumulacija izotopa se detektuje 3-16 dana prije pojave infarktne zone (smanjenje signala izotopa) na kompjuterizovanom tomogramu mozga. MRI mozga pokazuje lezije koje se pretežno nalaze u okcipitalnim režnjevima, koje mogu biti prolazne. Pretežno je zahvaćen okcipitalni korteks, u manjoj mjeri oštećena je bijela tvar. Na T2-ponderisanim slikama, lezije mozga u MELA se pojavljuju kao područja povećanog intenziteta signala. Brojni autori povezuju prolazna hiperintenzivna područja s reverzibilnim vaskularnim edemom.
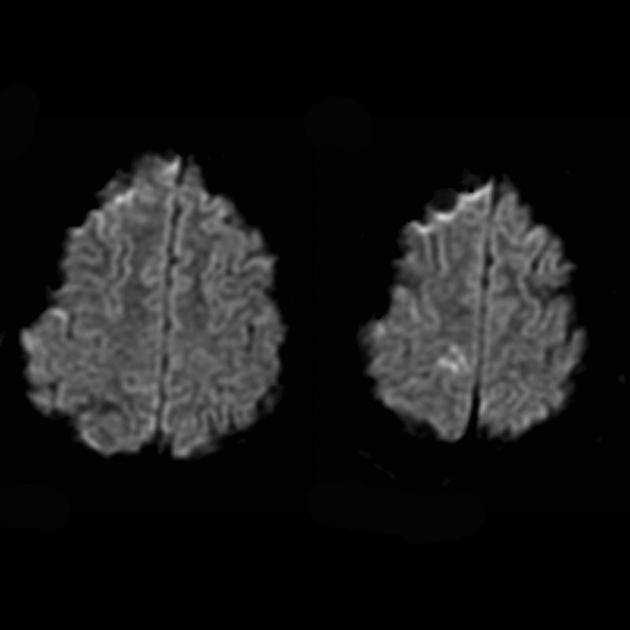
Angiografija obično ne otkriva vaskularne abnormalnosti. Difuzijsko ponderisana MRI pokazuje promjene povezane s vazogenim edemom.
Histopatologija: Biopsija mišića otkriva vlakna sa pokidanim "crvenim ivicama". Autopsiju mozga karakterizira kombinacija starih i novih žarišta infarkta, kao i atrofija korteksa sa žarišnim žarištima nekroze.
Trenutno je terapija suportivna. Glavni smjer liječenja je poboljšanje energetske ravnoteže mitohondrija i respiratornog lanca. Primijeniti koenzim p10 (80-300 mg / dan), vitamine K1 i KZ (25 mg / dan), jantarnu kiselinu (do 6 g / dan), vitamin C (2-4 g / dan), riboflavin (100 mg / dan). dan) i nikotinamid (do 1 g/dan). U vezi sa razvojem sekundarnog nedostatka karnitina, pacijentima se propisuje L-karnitin (do 100 mg/kg/dan). Vitamin E (300-500 mg/dan) i vitamin C (2-4 mg/dan) se koriste kao antioksidativna terapija.
Ne postoje opšteprihvaćeni režimi antiepileptičke terapije za MELA. Brojni autori predlažu da se isključe lijekovi koji mogu inhibirati energetski metabolizam (barbiturati, lijekovi valproične kiseline; kao i neki lijekovi iz drugih grupa, na primjer, hloramfenikol). U literaturi je opisano nekoliko izolovanih slučajeva pogoršanja napadaja uz upotrebu valproične kiseline u MELA sindromu sa mutacijom A3243C. Glavni AED lijekovi u liječenju epilepsije kod MELA sindroma su tegretol (ili trileptal), topamax, kepra u prosječnim terapijskim dozama. Pravilno odabrana terapija dovodi do značajnog smanjenja učestalosti sekundarnih generaliziranih konvulzivnih napadaja. Međutim, napadi s oštećenjem vegetativno-visceralne i vizualne funkcije obično su otporni na liječenje. U terminalnoj fazi bolesti, učestalost epileptičkih napadaja može se smanjiti.
Ovo je istorijat slučaja pacijenta sa dijagnozom MELAY sindroma verifikovanog tokom njegovog života.
Pacijent Ch.A., star 11 godina, posmatran je u Centru za dječju neurologiju i epilepsiju. Prilikom prijema pritužbe su se javljale na postupni gubitak govornih vještina, izražen poremećaj hoda s odbijanjem hodanja, značajno smanjenje vida, hirovitost i negativno ponašanje; svakodnevni serijski napadi u vidu trzanja mišića lica, mišića gornjih i donjih ekstremiteta, kao i kratkotrajne epizode gubitka vida.
Debi bolesti zabilježen je u dobi od 5 godina i 9 mjeseci. Prvi put, na pozadini punog zdravlja, prilikom uspavljivanja pojavila se jaka glavobolja, jednostavne vizualne halucinacije ("žuti zrak"), praćene nasilnim okretanjem očiju i glave u stranu i razvojem generaliziranog toničko-klonički konvulzivni napad, nakon čega je zabilježeno povraćanje. Nakon 9 mjeseci napadi sa istim simptomima su se ponavljali i brzo dobijali serijski karakter. Nakon imenovanja tegretola u dozi od 400 mg dnevno, učestalost napada se smanjila na 1 put mjesečno. Tegretol je zamijenjen Depakine Chrono u dozi od 900 mg/dan, protiv čega je zabilježena klinička remisija u trajanju od 6 mjeseci. S obzirom na klinički simptom
TOM IV BROJ 3 2009
tomatika, ograničenje napadaja na period uspavljivanja, normalna inteligencija bolesnika, pozitivna reakcija na valproat, dijagnosticirana je idiopatska okcipitalna epilepsija.
U dobi od 7 godina, fokalni verzivni napadaji nastavljeni su sa sekundarnom generalizacijom pri uspavljivanju sa istom učestalošću od 1 puta mjesečno. Povećanje doze Depakina na 1500 mg/dan nije dovelo do smanjenja učestalosti napadaja. Kada je lamiktal dodat u dozi od 75 mg/dan, napadi su prestali 4 mjeseca, a zatim su se nastavili istom učestalošću. U dobi od 8 godina pridružili su se napadi s kratkotrajnim gubitkom vida. Od 8 godina 8 mjeseci prije uspavljivanja počeli su se pojavljivati atipični izostanci: brzo treptanje sa zatvaranjem očnih kapaka i postavljanjem očnih jabučica prema gore; svest fluktuira.
U dobi od 9 godina pojavili su se višestruki serijski napadi, koji su trajali po nekoliko dana, sa jednostavnim vizualnim halucinacijama u vidu bljeskajućeg "zraka" ispred očiju, sa okretanjem očiju i glave udesno. Prije uspavljivanja, takvi napadi su ponekad prelazili u fokalne hemiklonske, koji su se manifestirali smanjenjem lica.
muskulatura desno, trzanje glave udesno, klonije desnih udova (veće od ruke). Ponekad se nakon napada javlja jaka glavobolja i povraćanje. U istoj dobi javljaju se inhibicijski napadi: aura u vidu naježivanja u nožnom palcu desne noge, praćena kratkotrajnom slabošću desne noge i nespretnošću desne ruke. Topamax je uveden u režim liječenja u dozi od 100 mg/dan - nije bilo epileptičkih napada 1 godinu.
Takođe, u dobi od 9 godina prvi put su se javila paroksizmalna stanja, praćena jakom glavoboljom, povraćanjem i razvojem hemipareze na desnoj strani. U nekim slučajevima ovakva stanja su bila praćena amaurozom koja je trajala od nekoliko minuta do nekoliko dana.
U dobi od 10,5 godina ponovo se javljaju napadi u vidu okretanja glave ulijevo, naglih pokreta očnih jabučica ulijevo, u trajanju do 5 s, učestalosti do 3 puta na sat, dnevno, čak i tokom spavanja. Doza Topamaxa je povećana na 150 mg/dan bez značajnog efekta. Sa 10 godina i 10 mjeseci. nakon intenzivne glavobolje, naizmjenično
Rice. 1. Pacijent Ch.A. 10 godina. Dijagnoza: MEAE sindrom. Simptomatska fokalna epilepsija.
Video-EEG monitoring (2004): u pozadini difuznog usporavanja glavne aktivnosti mozga, kontinuirana epileptiformna aktivnost se bilježi u lijevoj okcipitalnoj regiji. Subklinički EEG obrasci napada su takođe registrovani u lijevoj okcipitalnoj regiji sa širenjem na lijevu stražnju temporalnu regiju.
Centar za dječju neurologiju i epilepsiju
pod rukovodstvom profesora K.Yu. Mukhina se bavi dijagnostikom i liječenjem anksioznih poremećaja nervnog sistema u Aeteiju, specijalizirana je za etske oblike epilepsije.
Glavni pravci
aktivnosti:
Epilepsija kod djece i adolescenata
Glavobolja
Poremećaji spavanja kod djece
Tiki, enureza
Pregledi djece u prvim ^ mjesecima života.
Pregledi u našem centru:
Dijagnostika i liječenje bolesti nervnog sistema kod djece
Potpuna dijagnostika (uključujući predhiruršku) i liječenje epilepsije
Konsultacije neurologa i epileptologa
Konsultacije pedijatra (često bolesna djeca, gastroenterolog itd.)
Konsultacije psihijatra i psihologa.
Genetske konsultacije sa testovima (uključujući kariotipizaciju)
Video-EEG praćenje (u posebno opremljenim prostorijama Centra ili uz posjetu domu pacijenta)
Kompjuterska (digitalna) elektroencefalografija
UZDG (ultrazvučna doplerografija) krvnih sudova glave i vrata
Ehoencefalografija (ECHO EG)
Na našoj stranici možete se pretplatiti na časopis "Ruski časopis dječje neurologije" putem interneta.
Detaljne informacije o radu Centra od 10:00 do 19:00 sati na telefon:
Tel.: (+7495) 983-09-03; (+7926)290-50-30 Tel./Fax: (+7495) 394-82-52
Adresa: st. Borisovskie Prudy, 13, bl. 2. Internet: www.epileptologist.ru E-mail: [email protected](za detaljnu kartu rute pogledajte web stranicu)
TOM IV BROJ 3 2009
fokalni hemklonični i sekundarno generalizirani napadi koji su postali serijski i trajali 48 sati. Frizium je dodan u topamax u dozi od 10 mg/dan sa privremenim pozitivnim efektom.
Od 8. godine počele su se uočavati poteškoće s asimilacijom školskog materijala; smanjena memorija. Pojavio se povećan umor, iscrpljenost, inhibicija mentalne aktivnosti. Dječak je postao hirovit, razdražljiv, negativan; pozadina raspoloženja se smanjila. Od 9. godine došlo je do povećanja ove simptomatologije.
Iz anamneze života poznato je da je dijete rođeno iz druge normalne trudnoće, drugog termina, porođajne težine 2800 g, dužine 53 cm.Rani psihomotorni i govorni razvoj bio je u potpunosti usklađen sa godinama. Prethodne bolesti: vodene boginje sa 6 godina, česte akutne respiratorne virusne infekcije (do 4 puta godišnje) od 6 godina. Nasljednost za epilepsiju i druge neurološke bolesti nije opterećena.
U vrijeme pregleda (11 godina) stanje djeteta je bilo teško; negativno reaguje na inspekciju. Svjestan, pro-orijentisan
prostor i vrijeme. U kontakt stupa krajnje nevoljko, odbija slijediti upute. Spontani nistagmus ulijevo, glava nagnuta na lijevo rame sa okretom udesno. Jezik je u srednjoj liniji, faringealni refleks je smanjen; Primjećuju se disfagija i dizartrija. Vid je smanjen.
Određuje se umjerena difuzna mišićna hipotonija. Tetivni refleksi su ravnomjerno smanjeni. Došlo je do blagog smanjenja mišićne snage na desnim udovima. Patološki refleksi stopala nisu otkriveni. Ne postoje objektivni podaci o povredi osjetljivosti. Ne isplati se na Rombergovom testu. Odbija da hoda. Kada pokušate da ga dignete na noge, on plače, sjeda na pod. Nedostaje prilikom izvođenja testa indeksa prsta. Govori polako, pojedinačnim rečima, nevoljko.
Dodatne metode ispitivanja. Video-EEG monitoring (2004). Značajno usporavanje glavne aktivnosti snimanja u pozadini. Tokom studije zabilježena je kontinuirana epileptiformna aktivnost u lijevoj okcipitalnoj regiji sa širenjem na lijevu stražnju temporalnu regiju i sa periodičnim formiranjem EEG obrasca.
rođen 1993 16/12/05
Rice. 2. Pacijent Ch.A. 11 godina. Dijagnoza: MELAS sindrom. Simptomatska fokalna epilepsija.
Video-EEG praćenje je provedeno u dinamici nakon 1 godine (2005): značajno usporavanje pozadinske aktivnosti mozga. Prilikom snimanja u snu, kontinuirano regionalno usporavanje se bilježi u desnoj fronto-centralnoj regiji, u čijoj strukturi se detektuje vršna talasna aktivnost u desnoj fronto-centralnoj regiji.
ORIGINALNI ČLANCI
stupa (sl. 1). Takođe, utvrđeno je nastavak regionalnog usporavanja u desnoj fronto-centralnoj regiji sa uključivanjem pojedinačnih oštrih talasa.
Video-EEG praćenje u dinamici (2005): Značajno usporavanje pozadinske aktivnosti mozga. Studija je zabilježila nastavak regionalnog usporavanja u desnoj fronto-centralnoj regiji. U strukturi regionalnog usporavanja u desnom fronto-centralnom području otkriva se vršno-valna aktivnost (sl. 2).
MRI mozga. Prva magnetna rezonanca (6 godina) otkrila je jedan hiperintenzivan signal u T2 modu u lijevoj hemisferi malog mozga. MRI studija tokom vremena (10,5 godina): otkriveno je značajno pogoršanje primarne lezije sa širenjem patološkog procesa na lijevu i desnu okcipitalno-parijetalnu regiju obje hemisfere mozga (profesor A.A. Alikhanov).
Vizuelni evocirani potencijali: značajne morfološke i funkcionalne promene u vizuelnom aferentnom sistemu na nivou očnog nerva i kortikalnog dela vizuelnog analizatora, izraženije sa leve strane.
Konsultacije oftalmologa: djelomična atrofija očnih živaca. Elementi kortikalne agnozije.
Elektrokardiogram: ektopični ritam sa ubrzanjem do 100 otkucaja u minuti.
Vertikalni položaj električne ose srca. Promjene u repolarizacijskim procesima, koje su izraženije kod ortostaze.
Elektroneuromiografija: otkriva primarni mišićni tip lezije. Brzine provodljivosti duž perifernih nerava se ne smanjuju.
Proučavanje nivoa laktata u krvi: sadržaj laktata u krvi je 3,0 mmol / l (norma je do 1,8).
Uzimajući u obzir prisustvo epileptičkih napada koji potiču iz okcipitalnih regija korteksa velikog mozga, otpornih na terapiju, epizode nalik na moždani udar, periode amauroze, kognitivnog opadanja, prisustvo hiperintenzivnih signala u malom mozgu i stražnjim regijama moždane kore na Predložena je magnetna rezonanca, povećanje nivoa laktata u krvi, pacijentu je postavljena dijagnoza MELAS sindroma. Tokom genetskog pregleda, u krvnim ćelijama je pronađena mutacija A3243G u heteroplazmatskom stanju (dijagnostika je postavljena u Moskovskom državnom istraživačkom centru Ruske akademije medicinskih nauka), a dijagnoza je verifikovana.
Posmatranjem u praćenju utvrđeno je brzo napredovanje poremećaja viših mentalnih funkcija, razvoj kortikalne sljepoće, potpuna nepokretnost bolesnika, praćena nastankom smrti u dobi od 12 godina i 10 mjeseci. (nakon 7 godina od pojave bolesti).
Bibliografija
1. Nikolaeva E.A., Temin P.A. Mitohondrijalne bolesti praćene poremećenim neuropsihičkim razvojem. MELAS sindrom // Nasljedni poremećaji neuropsihičkog razvoja djece. Vodič za doktore koji je uredio Temin P.A. Kazantseva L.Z. - Medicina, 2001. - S. 96-107.
2. Nikolaeva E.A., Temin P.A., Nikanorova M.Yu., Klembovsky A.I., Suhorukov V.S., Dorofeeva M.Yu., Korsunsky A.A. Liječenje djeteta s mitohondrijskim sindromom MELAS (mitohondrijalna encefalopatija, laktacidoza, epizode slične moždanom udaru) // Ruski bilten za perinatologiju i pedijatriju. - 1997. - br. 2. - S. 30-34.
3. Smirnova I.N., Kistenev B.A., Krotenkova M.V., Suslina ZA. Tijek mitohondrijske encefalomiopatije sličan moždanom udaru (MELAS sindrom) // Atmosfera. Nervne bolesti. - 2006. - br. 1. - S. 43-48.
4. Temin PA, Nikanorova M.Yu., Nikolaeva E.A. MELAS sindrom (mitohondrijalna encefalomiopatija, laktacidoza, epizode slične moždanom udaru): glavne manifestacije, dijagnostički kriteriji, mogućnosti liječenja // Nevrol. časopis - 1998. - br. 2. - S. 43-48.
5. Ajmone-Marsan C., Ralston B. Epileptički napad, njegova funkcionalna morfologija i dijagnostički značaj. - Springfield (IL): Charles C. Thomas, 1957. - P. 3-231.
6. Aldrich M.S., Vanderzant C.W., Alessi A.G., Abou-Khalil B., Sackellares J.C. Iktalno kortikalno sljepilo s trajnim gubitkom vida // Epilepsija. - 1989. - V. 30. - P. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. Slučaj MELAS-a koji predstavlja složeni parcijalni epileptični status // Rinsho Shinkeigaku. - 2001. - V. 41(8). - P. 487-90.
TOM IV BROJ 3 2009
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptik fenotipovi povezani s mitohondrijskim poremećajima // Neurologija. - 2001. - V. 56(10). - P. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. Valproična kiselina pogoršava epilepsiju uzrokovanu MELAS-om kod pacijenta s A3243G mutacijom mitohondrijske DNK // Metab Brain Dis. - 2007. - V. 22(1). - str. 105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. et al. Molekularna patologija MELAS-a i MERRF-a. Odnos između opterećenja mutacijom i kliničkih fenotipova // Mozak. - 1997. - V.120. - P. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Polimorfizam epilepsije povezan s A3243G mutacijom mitohondrijalne DNK (MELAS): razlozi odgođene dijagnoze // Rev Neurol (Paris). - 2004. - V. 160(8-9). - P. 824-829.
12. Dvorkin G., Andermann F., Carpenter S. Klasična migrena, nepopravljiva epilepsija i višestruki moždani udari: sindrom povezan s mitohondrijalnom encefalopatijom / U: Andermann F., Lugaresi E., urednici. migrene i epilepsije. - Boston: Butterworths, 1987. - P. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Nalazi serijskih elektroencefalografa kod pacijenata s MELAS-om // Pediatr Neurol. - 1999. - V. 20(1). - str. 43-48.
14. Goto Y., Nonaka I., Horai S.A. Mutacija tRNA leu(UUR) gena povezana s MELAS podgrupom mitohondrijalnih encefalomiopatija // Priroda. - 1990. - V. 348. - P. 651-653.
15. Hasuo K., Tamura S., Yasumori K., Uchino A., Goda S., Ishimoto S., et al. Kompjuterizirana tomografija i angiografija u MELAS-u (mitohondrijska miopatija, encefalopatija, laktacidoza i epizode slične moždanom udaru): izvještaj o 3 slučaja // Neuroradiologija. - 1987.-V. 29. - P. 393-397.
16. Hirano M., Pavlakis S.G. Mitohondrijalna miopatija, encefalopatija, laktacidoza i epizode slične moždanom udaru (MELAS): Aktuelni koncepti // J. clin. Neurol. - 1994. - V. 9. - P. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto, Hirose G. Epileptički napadi kod pacijenata sa mitohondrijalnom miopatijom, encefalopatijom, mliječnom kiselinom i epizodama sličnim moždanom udaru ( MELAS) // Jpn J Psychiatry Neurol. - 1989. - V. 43(3). - P. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J., et al. Mitohondrijska encefalomiopatija s elevacijom laktata-piruvata i infarktom mozga // Neurologija. - 1984. - V. 34. - P. 72-77.
19. Kuzniecky R. Simptomatska epilepsija okcipitalnog režnja // Epilepsija. - 1998. - V. 39 Suppl 4. - P. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Dubina i direktno kortikalno snimanje kod poremećaja napadaja ekstratemporalnog porijekla // Neurology. - 1976. - V. 26. - P. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Klinički iktalni obrasci u epileptičkih pacijenata s okcipitalnim elektroencefalografskim žarištima // Neurology. - 1975. - V. 25. - P. 463-471.
22. Matthews P.M., Tampieri D., Berković S.F., Andermann F., Silver K., Chityat D., et al. Magnetna rezonanca pokazuje specifične abnormalnosti u MELAS sindromu // Neurologija. - 1991. - V. 41. - P. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. Slučaj MELAS-a povezan s epilepsijom partialis continua // No To Hattatsu. - 1991. - V. 23(1). - str. 65-70.
24. Montagna P., Gallassi R., Medori R., Govoni E., Zeviani M., Di Mauro S., et al. MELAS sindrom: karakteristične migrene i epileptičke karakteristike i prijenos na majku // Neurologija. - 1988. - V. 38. - P. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N., et al. Cerebralni protok krvi u mitohondrijalnoj miopatiji, encefalopatiji, laktacidozi i epizodama sličnim moždanom udaru // Moždani udar. - 1993. - V. 24. - P. 304-309.
26. Pavlakis S.G., Phillips P.C., Di Mauro S. et al. Mitohondrijska miopatija, encefalopatija, laktacidoza i epizode slične moždanom udaru: prepoznatljiv klinički sindrom // An neurol. - 1984. - V. 16. - P. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. Karakteristike epileptičnog statusa u MELAS-u. Analiza četiri slučaja // Neurologia. - 2006. - V. 21(1). - str. 1-11.
28. Williamson P.D., Spencer S.S. Kliničke i EEG karakteristike kompleksnih parcijalnih napadaja ekstratemporalnog porijekla // Epilepsija. - 1986. - V. 27 (Suppl 2). - str. 46-63.
29. Williamson P.D., Thadani V.M., Darcey T.M., Spencer D.D., Spencer S.S., Mattson R.H. Epilepsija okcipitalnog režnja: kliničke karakteristike, obrasci širenja napadaja i rezultati operacije // Ann Neurol. - 1992. - V. 31. - P. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Paradoksalni učinak natrijevog valproata koji pogoršava epilepsiju MELAS-a kod pacijenta sa A3243G mutacijom mitohondrijske DNK // Central European Journal of Medicine. - 2007. - V. 2(1). - P.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. Vasogeni edem na MELAS-u: serijska studija sa difuzijsko-ponderisanim MR slikanjem // Neurology. - 1999. - V. 53. - P. 2182-2184.
MELAS sindrom (MELAS) je prvi put opisao S. Pavlakis i kolege 1984. godine. Ali neki istraživači vjeruju da je sindrom već ranije označen konceptima kao što su obiteljska poliodistrofija, mliječna acidemija.
Suština patologije
Godine 1994. S. Pavlakis i Mizio Hirano opisali su 110 slučajeva bolesti. MELAS (Mitohondrijska encefalomiopatija, laktična acidoza i epizode slične moždanom udaru) je multisistemska progresivna neurodegenerativna bolest. Polimorfna je i karakterizirana je encefalopatijom sa konvulzijama i demencijom, laktacidozom. Bolest je uzrokovana mutacijama u mitohondrijskoj DNK (mtDNK). Ova bolest ima drugo ime - mitohondrijalna encefalomiopatija.
Opće informacije
Od 25 do 44% slučajeva bolesti je nasljedno, prenosi se po majčinoj liniji. U drugim slučajevima javlja se prvi put. Trenutno je poznato više od 10 gena koji mutiraju i dovode do razvoja ovog sindroma. To su geni koji kodiraju funkcije prijenosne RNK. MELAS sindrom se odnosi na mitohondrijalne bolesti (MD) sa abnormalnom akumulacijom mitohondrija, što dovodi do poremećaja cjelokupnog energetskog metabolizma ćelije.
Bolesti ove grupe prenose se samo po majčinoj liniji. Njima se u različitim kombinacijama zahvaćaju energetski najzavisniji organi i tkiva: skeletni mišići, srce, mozak, vid, jetra i bubrezi.
Simptomi su polimorfni i mogu se pojaviti u bilo kojoj dobi. Uključuje manifestacije dijabetesa, gubitak sluha, napade, endokrinopatije, nizak rast, srčane patologije, apsolutnu nemogućnost vježbanja i psihomotorne abnormalnosti.
Prije toga, psihomotorni razvoj je potpuno normalan. Nisu identifikovani pacijenti sa istim simptomima, jer mutacija utiče na mnoge gene: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTTS2, MTND1, 5, 6. Njihov broj nastavlja da raste.
Kod 80% pacijenata, MELAS sindrom je uzrokovan tačkom supstitucije A3243G u leucinskom tRNA genu (UUR).
Učestalost je nepouzdano proučavana. Postoji samo nekoliko podataka: na primjer, u Finskoj, stopa mutacije A3243G iznosila je 16:100 hiljada populacije; u Engleskoj - 1 slučaj na 13 hiljada ljudi.
Patološke promjene

Karakteristična patološka karakteristika MELAS sindroma su raščupana crvena vlakna (RRF), koja se mogu vidjeti u mišićnom tkivu sa posebnom Gomory trobojkom. Oni su rezultat mutiranih gena i morfološkog supstrata oštećenja mtDNK, nastalih kao rezultat proliferacije ovih abnormalnih mitohondrija.
Šta je uopšte mitohondrija
Mitohondrije su dvomembranska organela eukariotske ćelije (ćelije koja ima jezgro), čija je glavna funkcija opskrba energijom. Naime, mitohondrije su energetska baza ćelija, njihove energetske stanice.
Broj mitohondrija u ćelijama može varirati tokom njenog života od nekoliko do hiljada. I više ih se događa u ćelijama povezanim s proizvodnjom energije.
Same mitohondrije su najčešće okruglo izdužene, veličine od 1 do 10 mikrona. Mogu se zamrznuti nepomično ili se kretati unutar citoplazme ćelije. Obično se kreću tamo gdje je potrebno više energije.
Na unutrašnjoj membrani mitohondrija nalaze se izrasline (kriste) na kojima se nalaze čitavi sistemi enzima. U osnovi, to su proteinska jedinjenja. Broj krista zavisi od intenziteta procesa sinteze. Na primjer, u mitohondrijima mišićnih stanica uvijek ih ima puno.
Mitohondrije imaju autonomni sistem sinteze proteina - DNK, RNK i ribozoma. Neki od potrebnih proteina sintetiziraju sami mitohondriji - 5%, a dio se dobija iz citoplazme - 95%. Energija se ekstrahuje iz organskih jedinjenja kroz različite enzimske reakcije.
Neke od ovih reakcija odvijaju se uz sudjelovanje kisika, odnosno dolazi do oksidacije, a nakon drugih se oslobađa CO 2 uz prijenos protona vodika i oslobađanje energije. Drugim riječima, mitohondrij je aktivan učesnik u ćelijskom disanju.
Ove reakcije se javljaju na kristama ili u samim mitohondrijama, što je toliko važno za ćeliju da će, ako se izliječi, stanica biti potpuno zdrava.
Patogeneza

Na prvi pogled, kod MELAS sindroma stanje podsjeća na varijantu nakon moždanog udara. Ali u stvari, to je netipično: javlja se kod mladih ljudi, često izazvano zaraznim bolestima, a može se javiti u obliku maligne glavobolje nalik migreni, konvulzija.
Angiografija ne daje nikakve vaskularne patologije. Mogu postojati normalni krvni sudovi ili povećani kalibri nekih arterija, vena ili se javlja kapilarna hiperemija.
MRI pokazuje da akutno oštećenje mozga kod MELAS sindroma može migrirati, pa čak i nestati. Neki fokusi fluktuiraju. Za tipičan moždani udar to je potpuno nekarakteristično.
Kod MELAS sindroma prisutna je multifokalna nekroza. Uglavnom, to je uočljivo u okcipitalnom dijelu (posteriorna lokalizacija) kore velikog mozga i bijeloj tvari subkorteksa. Ali mogu se pojaviti iu drugim dijelovima mozga. Ova područja podsjećaju na nekrozu kod srčanog udara, ali se nalaze izvan bazena centralnih cerebralnih žila.
Simptomatske manifestacije

Obično se MELAS sindrom kod djece javlja u dobi od 6-10 godina (može početi sa 3 godine i 40 godina). Rani početak bolesti je tipičniji i pogađa 90% pacijenata. Sa ranim početkom, bolest teče teže. Pacijenti su obično niski, slabog mišića i apsolutno neprilagođeni fizičkim naporima.
Svaka napetost ili fizička aktivnost čini da se osjećate gore. Od unutrašnjih organa, srce je zahvaćeno pothranjenošću mišića i provodljivosti, praćeno razvojem kardiovaskularne insuficijencije. Javljaju se i nefropatija, dijabetes, gastrointestinalne smetnje sa povraćanjem, smanjen je sluh. Karakteriziraju ga bol u mišićima, nedostatak refleksa, pareza, konvulzije, IPE, gubitak svijesti. Slabost mišića (miopatski sindrom) i senzorneuralni gubitak sluha su također tipični za ovu patologiju.
Endokrinopatiju predstavlja ne samo dijabetes melitus, već i nedostatak hormona rasta. Srčani i bubrežni poremećaji su rijetki u razvoju dotične bolesti.
Napadi kod MELAS sindroma su vrlo varijabilni. Mogu biti žarišne, generalizirane, toničko-kloničke i mioklonične. Karakteristična je apsolutna neosjetljivost napadaja na antikonvulzivnu terapiju. Često se dešava da lekari dijagnostikuju epilepsiju i prepišu, na primer, valproičnu kiselinu. Nakon nje, zdravstveno stanje se naglo pogoršava, a grčevi se povećavaju, jer depresira mitohondrije. Iako se demencija razvija, rijetko postaje manifestni simptom.
Također je karakterističan za bolest, ali se javlja i kod mnogih drugih patologija, pa stoga ne može poslužiti kao osnova za dijagnozu. Samo u kombinaciji s migrenama, konvulzijama i/ili pojavama sličnim moždanom udaru može se posumnjati na pojavu MELAS sindroma. Čak i ovako opsežni simptomi ne daju ispravnu dijagnozu. Progresija procesa se odvija na različite načine.
znakovi

Karakteristična klinička karakteristika MELAS sindroma su epizode slične moždanom udaru (IPE), u kojima se neurološki simptomi iznenada pojavljuju. IPE karakterizira asimetrija lezija. Oni mogu biti višestruki.
Selektivnost takve lokalizacije također daje određene žarišne simptome:
- hemianopsija (kortikalno sljepilo);
- hemipareza;
- senzorna afazija (nerazumijevanje riječi);
- akalkulija (kršenje računa);
- agrafija (pravopisne povrede);
- ataksija (poremećena koordinacija voljnih pokreta);
- promene u svesti.
Nije neuobičajeno da se ovi simptomi slični moždanom udaru vraćaju svakih 1 do 3 mjeseca. Posebnost akutnih epizoda u MELAS-u je da se brzo povlače, ali se često ponavljaju, odnosno kao da prolaze bez traga. Osim toga, kod pacijenata sa ovom bolešću, kalcifikacije se talože u bazalnim ganglijama (ovo se nalazi na CT).
Epizode slične moždanom udaru često se razvijaju u dobi od 5-15 godina. Nikada ne postaju rezultat tromboembolije. Angiopatija u MELAS-u je posljedica hiperproliferacije istih mitohondrija.
IPE se u simptomima manifestuje rekurentnim napadima cefalalgije, vrtoglavica, pareza, paraliza udova, kranijalnih nerava. Čovjek je potpuno demoralisan.
Laktacidoza u MELAS sindromu

Njegov glavni krivac je višak mliječne kiseline u krvi i tkivima nervnog sistema. Ovo naglo smanjuje kiselost krvi u arterijama. Takva acidoza je čest pratilac dijabetes melitusa, koji je prisutan kod MELAS sindroma.
U ranoj fazi, manifestacije su nespecifične. Uočavaju se sljedeći simptomi: opća slabost, bol u grudima, apatija, pospanost. Vrlo su karakteristični mijalgija nakon fizičkog napora i povremeno ubrzano disanje bez ikakvog mirisa.
U srednjem stadijumu dolazi do nakupljanja mliječne kiseline i javlja se hiperventilacijski sindrom (HVS). Ugljični dioksid se nakuplja u krvi. Počinje da se formira bučno disanje - Kussmaul. Pritisak pada do kolapsa, nastupa oligurija. Pacijent postaje nemiran, deliričan, a zatim gubi svijest s naknadnim razvojem kome - ovo je posljednja faza. Simptomi laktacidoze se razvijaju brzo, naime, u roku od nekoliko sati. Onda dolazi smrt.
Dijagnostičke mjere

Kao što je već napomenuto, zbog polimorfizma simptoma i mutacije velikog broja gena, dijagnoza MELAS sindroma je teška. Drzati:
- opći i biohemijski testovi krvi;
- biopsija mišića;
- genetska studija sa uporednom analizom među bolesnim srodnicima;
- CT mozga: područja infarkta češće u hemisferama, rjeđe u malom mozgu, bazalnim ganglijama;
- povećanje kalibra krvnih žila (arterije, vene, kapilare);
- DNK dijagnostika: traženje karakterističnih tačkastih mutacija u mtDNK.
Terapijske metode
Liječenje MELAS sindroma nije razvijeno i trenutno je neizlječivo. Postoje samo pokušaji da se uspori proces poraza. Liječenje ide u dva smjera: postsindromska terapija (epilepsija, dijabetes melitus) i patogenetska. Međutim, danas ne postoji efikasna patogenetska terapija.
Postoje simptomatski tretmani: kod gubitka sluha aktivno se koriste slušni aparati, kod slabosti respiratornih mišića pruža se respiratorna terapija. Uočeno je da je kod MELAS sindroma u krvi pacijenata nivo L-arginina značajno smanjen tokom IPE. Stoga se terapija provodi preparatima arginina i vitaminima. Proučava se pozitivno djelovanje koenzima Q ili idebinona (noben), preparata jantarne kiseline, vitamina K 1 i K 3, B 2 , B 3 , E, C; L-karnitin, antioksidansi (meksidol, mildronat), korektori laktacidoze (dimefosfon) - svi oni poboljšavaju energetski metabolizam ćelije. U liječenju napadaja, valproati i barbiturati se ne propisuju, jer depresiraju mitohondrije.
Kao prevenciju ovog sindroma, najbolje je pribjeći IVF metodi. Ako žena zna da ima slučaj ispoljavanja ove bolesti u svojoj porodici, onda se citoplazma za oplodnju uzima od zdrave žene. Metoda je još u fazi proučavanja, nije masovna.
Neke karakteristike
Dijagnoza mitohondrijalnih poremećaja zahtijeva vrlo pažljiv pristup terapiji. Sredstva za metaboličko djelovanje moraju nužno biti uključena u to. Stabiliziraju procese tkivnog disanja, oksidativne fosforilacije u stanicama. Samo sistematsko sprovođenje takvog tretmana može pomoći u održavanju stanja pacijenata, sprečavajući epizode moždanog udara.
Prognoza
Prognoza je nepovoljna zbog nedostatka efikasnog liječenja. Očekivano trajanje života od prve pojave simptoma obično ne prelazi pet godina. Nepoznavanje uzroka bolesti dovodi do toga da još uvijek nije pronađen optimalan režim liječenja. Sve to čini šanse za izlječenje minimalnim.
MELAS sindrom je mitohondrijski poremećaj karakteriziran zahvaćenošću mišića i CNS-a.
MELAS (eng. Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes - “mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes”) je progresivna neurodegenerativna bolest koju karakteriziraju manifestacije navedene u naslovu, a prate ga - pomorfni simptomi. moždani udar, dijabetes, napadi, smanjeni gubitak sluha, bolesti srca, nizak rast, endokrinopatije, netolerancija na vježbanje i neuropsihijatrijski poremećaji.
Priča.
MELAS sindrom su prvi put opisali Pavlakis i kolege 1984. godine; deset godina kasnije, Pavlakis i Mizio Hirano objavili su pregled 110 slučajeva.
vrsta nasljeđivanja:
majčinski
Epidemiologija:
Tačna učestalost bolesti nije poznata. U literaturi je malo podataka o učestalosti bolesti. U sjevernoj Finskoj, stopa mutacije A3243G je 16,3:100,000.
Patogeneza:
Mutacije mitohondralne DNK, koje kontrolišu respiratorni lanac mitohondrija, praćene su poremećajem u procesima oksidativne fosforilacije, najvažnijeg izvora energije za metaboličke procese u ćeliji.
Kliničke manifestacije
U dobi od 40 godina, pacijenti sa MELAS-om se primaju sa klinikom prolaznog ishemijskog napada, kao i sa epilepsijom, ponovljenim povraćanjem, glavoboljom i slabošću mišića. Ovim pacijentima se često klinički dijagnosticira demencija.
Mladost i odsustvo faktora rizika specifičnih za moždani udar pomažu razmišljanju o MELAS-u.
Laboratorijski podaci
Laktatna acidoza - povećani nivoi laktata i piruvata.
Podaci o vizualizaciji
Promjene u mozgu slične su promjenama kod moždanog udara.
Razlike od moždanog udara
1) zahvaćena područja se ne poklapaju sa granicama arterijskih vaskularnih bazena.
2) sa ponovljenim napadima, žarišta se vizualiziraju u različitoj lokalizaciji.
+ klinički podaci (mlada dob, bez faktora rizika za moždani udar).
CT
Višestruka hipodenzna područja koja nisu u skladu sa vaskularnim krevetom.
Kalcifikacija bazalnih ganglija (najčešća kod starijih pacijenata).
Atrofija se javlja u pozadini regresije i kliničkog poboljšanja.
MRI
Akutni infarkt
Za diferencijaciju sa moždanim udarom koriste se ADC i DWI (ograničenje difuzije kod moždanih udara (citotoksični edem), a kod MELAS-a difuzija je blago ograničena ili nepromijenjena (vazogeni edem).
Uključenost u patološki proces subkortikalne bijele tvari mozga.
Pogoršanje vizualizacije jasnoće kontura konvolucija i povećanje signala od njih na T2-ponderiranim slikama.
Hronični infarkt
Promjene mogu biti simetrične ili asimetrične.
Fokalna atrofija se javlja u pozadini regresije i kliničkog poboljšanja.
Najčešće su zahvaćeni parijetalni, okcipitalni i temporalni režanj mozga.
MR spektroskopija
Povećani nivoi laktata.




Materijali su namijenjeni neurolozima, terapeutima i liječnicima opće prakse.
Sergej Lihačov, šef, dr. nauke, profesor;
Inessa Pleshko, vodeći istraživač, dr. nauka, Neurološko odeljenje Republičkog naučno-praktičnog centra za neurologiju i neurohirurgiju.
Cerebralna autosomno dominantna arteriopatija sa subkortikalnim infarktom i leukoencefalopatijom (CADASIL) je progresivna autosomno dominantna bolest, čije kliničke manifestacije uključuju rekurentne subkortikalne ishemijske moždane udare, migrenu, subkortikalnu demenciju i afektivne poremećaje. Trenutna prevalencija - 1 slučaj
na 100.000 stanovnika.
Republički naučno-praktični centar za neurologiju i neurohirurgiju prima 7 pacijenata (od toga 4 žene) sa CADASIL-om; starost - od 32 do 68 godina. Ispitivani su neurološkim, molekularno genetskim metodama. Bilo je karakterističnih simptoma; u anamnezi - migrena, rekurentni lakunarni moždani udari i afektivni poremećaji. MRI mozga otkrio je subkortikalne infarkte i leukoencefalopatiju karakterističnu za CADASIL.
Kao rezultat molekularne genetičke dijagnostike, 2 osobe su imale heterozigotnu mutaciju gena Notch3 na 19. hromozomu, što uzrokuje CADASIL. Notch geni kodiraju transmembranske receptore uključene u ontogenezu ćelije. Kod CADASIL-a se u većini slučajeva utvrđuju misense mutacije, zbog kojih se mijenja struktura transmembranskog proteina i narušava njegove funkcije.
Patogeneza CADASIL-a nije potpuno jasna. Smatra se da je glavni faktor arteriopatija s progresivnom okluzijom malih perforirajućih žila bijele tvari mozga (što dovodi do kronične hipoperfuzije). Istovremeno se nalaze karakteristične granularne osmiofilne inkluzije koje uzrokuju proliferaciju komponenti bazalne membrane, zadebljanje srednje membrane i mehaničku kompresiju malih arterija. Kao rezultat, krvno-moždana barijera je oštećena - razvija se edem.
Dodatni patološki faktor je aktivacija astrocita u blizini vaskularnog zida. Oni oslobađaju endotel-1, uzrokujući vazokonstrikciju i poremećen protok krvi.
Sastav granularnih osmiofilnih inkluzija nije poznat. Pretpostavlja se da je protein Notch3 jedna od njihovih komponenti. U biopsijama kože pacijenata sa mutacijom Notch3, osmiofilne granule i degeneracija glatkih mišićnih ćelija mogu se otkriti i prije 20. godine života.
Klinička dijagnostika CADASIL-a:
- opterećena porodična istorija;
- razvoj prvih simptoma bolesti prije 50. godine života;
- prisutnost dva od sljedećih simptoma - migrene, ponavljajućih moždanih udara, poremećaja raspoloženja, subkortikalne demencije.
Vaskularni faktori rizika etiološki povezani s neurološkim simptomima treba isključiti. MRI pokazuje oštećenje bijele tvari moždanih hemisfera i odsustvo kortikalnih infarkta.
Pouzdana dijagnoza "CADASIL" potvrđuje se pozitivnim rezultatom molekularne genetičke dijagnoze ili otkrivanjem arteriopatije sa karakterističnim granularnim osmiofilnim inkluzijama u biopsiji kože ili mišića.
Najčešći simptomi CADASIL-a su prolazni ishemijski napadi i ishemijski moždani udar, uočeni kod skoro 85% pacijenata.
Karakterizira ih rekurentni tok, koji se manifestuje klasičnim sindromima lakunarnih moždanih udara i potpunom kliničkom remisijom nakon nekoliko dana ili sedmica.
Druga po učestalosti su kognitivna oštećenja (zapažena kod 60% pacijenata). Može početi u dobi od 35 godina, ponekad čak i prije ishemijskih epizoda. Otprilike 75% CADASIL pacijenata razvije demenciju. Prvi simptom je obično migrena; često se javlja prije 20. godine života i obično prethodi moždanom udaru.
Podaci o učešću srca u patološkom procesu kod CADASIL-a su kontradiktorni. L. Oberstein et al. (2003) su otkrili da je 25% pacijenata s dijagnozom CADASIL-a imalo anamnezu akutnog infarkta miokarda ili patologije Q-talasa na elektrokardiogramu. U drugoj studiji, Cumurciuc et al. (2006) nisu pronašli pozitivnu srčanu anamnezu kod 23 osobe sa Notch3 mutacijom.
Kliničke manifestacije CADASIL-a i cerebralne mikroangiopatije različite etiologije su slične - potrebna je diferencijalna dijagnoza.
U cilju pravovremenog određivanja CADASIL-a kod pacijenata i njihovih porodica, potrebno je pribjeći molekularno-genetskim metodama i/ili histološkim studijama.
MELAS sindrom
Mitohondrijska encefalomiopatija s laktacidozom i epizodama sličnim moždanom udaru (MELAS) je rijetka nasljedna bolest uzrokovana patologijom mitohondrijalnog genoma, poremećenim energetskim metabolizmom i funkcionisanjem energetski najzavisnijih organa i tkiva (CNS, srčani i skeletni mišići, oči, bubrezi, jetra, koštana srž, endokrini sistem). Velika varijabilnost kliničkih manifestacija MELAS sindroma i rijetka pojava predodređuju dijagnostičke poteškoće za praktičara.
U Republičkom naučno-praktičnom centru za neurologiju i neurohirurgiju posmatraju se 3 pacijenta (žena od 46 godina i njeni sinovi 24 i 23 godine) sa dijagnostikovanim MELAS sindromom. Obavljeni su klinički i neurološki pregledi, molekularno-genetička dijagnostika, MR mozga.
Svi su kratki; u anamnezi - simptomi mitohondrijalne patologije: senzorneuralni gubitak sluha, glavobolje nalik migreni, loša tolerancija vježbanja. Početak bolesti su generalizirani konvulzivni napadi. Kod 2 bolesnika prvi simptomi su se javili prije 20. godine života; javljali su se jedan za drugim epileptični napadi, epizode oštećenja vida sa prisustvom žarišta na neuroima u okcipitalnoj i temporalnoj regiji, povećanje nivoa laktata u krvi i likvoru. 1 osoba je imala umjereno smanjenje kognitivnih funkcija; prema ultrazvuku srca - hipertrofična kardiomiopatija; dijabetesa.
Molekularno genetska studija otkrila je multisistemske lezije tipične za MELAS, široku varijabilnost i različite stepene kliničkih manifestacija, što odgovara broju kopija mutanta A3243G u tRNA Leu(UUR) genu.
MELAS karakteriše majčinski tip nasljeđivanja, prisustvo sporadičnih slučajeva kada dođe do de novo mutacije; akumulacija u ćelijama - normalnih i mutantnih tipova - mitohondrijalne DNK (heteroplazmija) i nasumična distribucija tokom podele između ćelija kćeri (mitotična segregacija). Na genetskom nivou, uzrok MELAS sindroma je heteroplazmatsko preuređenje 3243A>G u genu tRNALeu(UUR) (otkriveno je 80% slučajeva).
Patogeneza bolesti još nije proučavana. Postoje 2 glavne teorije - "mitohondrijalna angiopatija" i "mitohondrijalna citopatija". Poznato je da lezija nalik na moždani udar ne odgovara vaskularnim zonama i da se proteže na okolna područja zbog pratećeg vazogenog edema uzrokovanog produženom epileptičnom aktivnošću. Kao što se sugerira, epizode slične moždanom udaru nastaju zbog neuralne hiperekscitabilnosti u ograničenom području mozga. Nastaje zbog mitohondrijalne disfunkcije u kapilarnim endotelnim stanicama, ili u neuronima, ili u astrocitima; depolarizira susjedne neurone, što dovodi do širenja epileptičke aktivnosti.
Osim toga, u intervalima između epizoda sličnih moždanom udaru, prema jednofotonskoj emisionoj kompjuterizovanoj tomografiji (SPECT), pacijenti sa MELAS-om imaju hipoperfuziju zadnjeg cingularnog korteksa, što ukazuje na poremećaj cerebralne hemodinamike.
Kršenje oksidativne fosforilacije, ruptura mitohondrijalnog respiratornog lanca doprinose prevlasti kataboličkog metabolizma i promjenama iz Krebsovog ciklusa u anaerobnu glikozu s akumulacijom laktata. Visok nivo potonjeg u CNS-u obično korelira s periodima neuroloških simptoma.
Glavni klinički znaci MELAS-a su epizode slične moždanom udaru, laktacidoza i prisustvo "pocijepanih crvenih vlakana" u uzorcima mišićne biopsije. Dodatne manifestacije mogu biti demencija, psihoza, epileptički napadi, glavobolje nalik migreni, ataksija, miopatija, kalcifikacija bazalnih ganglija na neuroimagingu, optička atrofija, retinopatija, gluvoća, dijabetes, crijevna pseudo-opstrukcija, kardiomiopatija.
Rana dob MELAS debija je od 5 do 20 godina, međutim, postoje zapažanja kasnog početka - u 5.–6. deceniji života. Postoje slučajevi kada je sindrom počeo nakon srčanih poremećaja.
Multisistemska priroda lezija u MELAS-u komplikuje kliničku dijagnozu.
Nasljedna priroda bolesti obavezuje se na provođenje molekularno-genetskih studija kako bi se postavila tačna dijagnoza.
i identifikuju druge pacijente - među srodnicima pacijenta.
Materijali su namijenjeni neurolozima, terapeutima i liječnicima opće prakse.
| Mitohondrijalna miopatija, encefalomiopatija, laktacidoza i epizode slične moždanom udaru | |
|---|---|
| Kalcifikacija bazalnih ganglija, cerebelarna atrofija, povećanje laktata; CT snimak osobe sa dijagnozom MELAS | |
| Specijalitet | neurologija |
genetika
Biopsija mišića osobe kojoj je dijagnosticiran MELAS, ali ne nosi poznatu mutaciju. (a) Gomorijeva modificirana trobojna boja pokazuje neka hrapava crvena vlakna (strelice). (b) Boja citokrom c oksidazom, koja pokazuje tip-1, blago obojena vlakna i vlakna tipa II, tamna vlakna i nekoliko vlakana sa abnormalnim mitohondrijalnim zbirkama (strelice). Imajte na umu da se citokrom c oksidaza negativna vlakna obično vide kod mitohondrijalne encefalopatije, laktacidoze i epizoda sličnih moždanom udaru (MELAS). (c) Bojenje sukcinat dehidrogenazom pokazuje nekoliko neravnih plavih vlakana i intenzivno bojenje u mitohondrijama krvnih sudova (strelica). (d) Elektronska mikroskopija pokazuje abnormalnu kolekciju mitohondrija sa parakristalnim inkluzijama (strelice), osmiofilnim inkluzijama (velike strelice) i mitohondrijalnim vakuolama (male strelice).
MELAS je uzrokovan mutacijama gena u mitohondrijskoj DNK.
NADH dehidrogenaze
Mutacije u MT-TL1 uzrokuju preko 80 posto svih slučajeva MELASA. Oni smanjuju sposobnost mitohondrija da stvaraju proteine, koriste kiseonik i proizvode energiju. Istraživači nisu utvrdili kako promjene u mitohondrijskoj DNK dovode do specifičnih znakova i simptoma MELAS-a. Oni nastavljaju da istražuju efekte mutacija mitohondrijalnih gena u različitim tkivima, posebno u mozgu.
nasljeđe
Ovo stanje se nasljeđuje u mitohondrijskom uzorku, koji je također poznat kao nasljeđe od majke i heteroplazmija. Ovaj obrazac nasljeđivanja odnosi se na gene sadržane u mitohondrijskoj DNK. Budući da jaja, ali ne i sperma, doprinose mitohondrijima embrionu u razvoju, samo ženke prolaze kroz mitohondrijalne uslove za svoje bebe. Mitohondrijski poremećaji mogu se pojaviti u svakoj generaciji porodice i mogu uticati i na muškarce i na žene, ali očevi ne prenose mitohondrijalne osobine svoje djece. U većini slučajeva, ljudi sa MELAS-om nasljeđuju izmijenjeni mitohondrijski gen od svoje majke. Ređe, poremećaj je rezultat nove mutacije mitohondrijalnog gena i javlja se kod ljudi bez porodične istorije MELAS-a.
dijagnostika
Liječenje / prognoza
Pacijentima se upravlja prema tome koja su područja tijela zahvaćena u određenom trenutku.