Hur länge lever barn med melas syndrom? Sällsynta sjukdomar. Symtom på MELAS-syndromet
Nyckelord
MELAS SYNDROM / MELAS SYNDROM / EPILEPSI / EPILEPSI / KLINIKanteckning vetenskaplig artikel om klinisk medicin, författare till vetenskapligt arbete - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
MELAS-syndromet är en genetiskt betingad sjukdom från gruppen mitokondriella sjukdomar, definierad som mitokondriell encefalomyopati med laktacidos och strokeliknande episoder (mitokondriell encefalomyopati, laktacidos med strokeliknande episoder). Alla organ och vävnader är involverade i den patologiska processen, men muskel- och nervsystemet lider i större utsträckning. Sjukdomen utvecklas oftast mellan 6 och 10 års ålder. Sjukdomsförloppet är progressivt. I de flesta fall manifesterar sjukdomen sig med epileptiska anfall, återkommande huvudvärk, kräkningar och anorexi. Epilepsi är en viktig klinisk manifestation av MELAS-syndromet. Epileptiska anfall är det första igenkännbara symtomet vid mitokondriell encefalopati (ME) i 53 % av fallen. Vid MELAS är occipital epilepsi den vanligaste. Med utvecklingen av sjukdomen noteras epilepsiresistens mot terapi, ofta med ett statusförlopp. Fall av transformation till Kozhevnikovs epilepsi beskrivs. Vi presenterar fallhistorien för en patient med diagnosen MELAS-syndrom verifierad under sin livstid.
Relaterade ämnen vetenskapliga artiklar i klinisk medicin, författare till vetenskapligt arbete - Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova S.V., Chadaev V.A.
-
Mitokondriell encefalopati med strokeliknande episoder och laktacidos (melas syndrom): diagnostiska kriterier, kännetecken för epileptiska anfall och tillvägagångssätt för behandling på exemplet på ett kliniskt fall
2017 / Yamin M.A., Chernikova I.V., Araslanova L.V., Shevkun P.A. -
Stroke vid mitokondriella sjukdomar
2012 / Pizova N.V. -
Epilepsi hos barn med mitokondriella sjukdomar: funktioner för diagnos och behandling
2012 / Zavadenko N. N., Kholin A. A. -
Neurologiska störningar vid mitokondriell encefalomyopati - laktacidos med strokeliknande episoder (MELAS-syndrom)
2012 / Kharlamov Dmitry Alekseevich, Krapivkin Alexey Igorevich, Sukhorukov Vladimir Sergeevich, Kuftina Lyudmila Andreevna, Groznova Olga Sergeevna -
Melas syndrom som en ovanlig orsak till hypoparatyreos: ett kliniskt fall
2018 / Umyarova Dilyara Shamilevna, Grebennikova Tatyana Alekseevna, Zenkova Tatyana Stanislavovna, Sorkina Ekaterina Leonidovna, Zhanna Belaya -
Strokeliknande episoder vid mitokondriell encefalomyopati med laktacidos
2010 / Kalashnikova Lyudmila Andreevna, Dobrynina L. A., Sakharova A. V., Chaikovskaya R. P., Mir-kasimov M. F., Konovalov R. N., Shabalina A. A., Kostyreva M. V., Gnezditsky V.V., Protsky S.V. -
Mitokondriella cytopatier: melas och MIDD-syndrom. En genetisk defekt, olika kliniska fenotyper
2017 / Muranova A.V., Strokov I.A. -
Benign occipital epilepsi i barndomen med en tidig debut (Panayotopoulos syndrom). Beskrivning av det kliniska fallet
2015 / Matyuk Yu.V., Kotov A.S., Borisova M.N., Panteleeva M.V., Shatalin A.V. -
Polymorfism av kliniska manifestationer av progressiv mitokondriell encefalomyopati associerad med POLG1-genmutation
2016 / Yablonskaya M.I., Nikolaeva E.A., Shatalov P.A., Kharabadze M.N. -
Diagnostiskt värde av studien av cytokemisk aktivitet hos enzymer i ärftliga mitokondriella sjukdomar
2017 / Kazantseva I.A., Kotov S.V., Borodataya E.V., Sidorova O.P., Kotov A.S.
EPILEPSI I MELAS SYNDROM
MELAS syndrom är en genetiskt betingad sjukdom i mitokondriella gruppen, definierad som mitokondriell encefalomyopati, laktacidos med strokeliknande episoder. Den patologiska processen involverar alla organ och vävnader, men den är mestadels negativ för muskel- och nervsystemet. Sjukdomen är vanligast hos barn i åldrarna 6 till 10. Det kliniska förloppet är progressivt. I de flesta fall manifesteras sjukdomen av epileptiska anfall, återfallande huvudvärk, kräkningar, anorexi. Den viktiga kliniska presentationen av MELAS syndrom är epilepsi. Epileptiska anfall är det initiala diagnostiseringssymptomet på mitokondriell encefalopati (ME) i 53 % av fallen. Occipital epilepsi är den vanligaste vid MELAS-syndrom. När sjukdomen fortskrider observeras epilepsiresistens mot behandling, ofta med uppkomsten av status epilepticus. Vissa fall av transformation till Kozhevnikovs epilepsi beskrivs.Anamnesen på en patient med en verifierad vid levande diagnos av MELAS-syndrom ges.
Texten till det vetenskapliga arbetet på ämnet "Epilepsi vid melas syndrom"
VOLYM IV NUMMER 3 2009
EPILEPSI MED MELAS SYNDROM
K.Yu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, C.B. Mikhailova2, VA. Chadaev1, AA. Alikhanov1-2, B.N. Ryzhkov1, A.S. Petrokhin1
EPILEPSI I MELAS SYNDROM
KYu. Mukhin1, M.B. Mironov1, N.V. Nikiforova1, S.V. Mikhailova2, UA. Chadaev1, AA. Alikhanov1-2, B.N. Ryzkov1 AS. Petrokhin1
1 - Institutionen för neurologi och neurokirurgi, fakulteten för pediatrik, statlig utbildningsinstitution för högre yrkesutbildning, Russian State Medical University of Roszdrav
2 - Ryska barnsjukhuset
MELAS syndrom är en genetiskt betingad sjukdom från gruppen mitokondriella sjukdomar, definierad som mitokondriell encefalomyopati med laktacidos och strokeliknande episoder (mitokondriell encefalomyopati, mjölksyra med strokeliknande episoder). Alla organ och vävnader är involverade i den patologiska processen, men muskel- och nervsystemet lider i större utsträckning. Sjukdomen utvecklas oftast mellan 6 och 10 års ålder. Sjukdomsförloppet är progressivt. I de flesta fall manifesterar sjukdomen sig med epileptiska anfall, återkommande huvudvärk, kräkningar och anorexi. Epilepsi är en viktig klinisk manifestation av MELAs syndrom. Epileptiska anfall är det första igenkännbara symtomet vid mitokondriell encefalopati (ME) i 53 % av fallen. Vid MELAS är occipital epilepsi den vanligaste. Med utvecklingen av sjukdomen noteras epilepsiresistens mot terapi, ofta med ett statusförlopp. Fall av transformation till Kozhevnikovs epilepsi beskrivs. Vi presenterar fallhistorien för en patient med diagnosen MELAS-syndrom verifierad under sin livstid.
Nyckelord: MELAS syndrom, epilepsi, klinik, diagnostik, behandling.
MELAS syndrom är en genetiskt betingad sjukdom i mitokondriella gruppen, definierad som mitokondriell encefalomyopati, laktacidos med strokeliknande episoder. Den patologiska processen involverar alla organ och vävnader, men den är mestadels negativ för muskel- och nervsystemet. Sjukdomen är vanligast hos barn i åldrarna 6 till 10. Det kliniska förloppet är progressivt. I de flesta fall manifesteras sjukdomen av epileptiska anfall, återfallande huvudvärk, kräkningar, anorexi. Den viktiga kliniska presentationen av MELAS syndrom är epilepsi. Epileptiska anfall är det initiala diagnostiseringssymptomet på mitokondriell encefalopati (ME) i 53 % av fallen. Occipital epilepsi är den vanligaste vid MELAS-syndrom. När sjukdomen fortskrider observeras epilepsiresistens mot behandling, ofta med uppkomsten av status epilepticus. Vissa fall av transformation till Kozhevnikovs epilepsi beskrivs.Anamnesen på en patient med en verifierad vid levande diagnos av MELAS-syndrom ges.
Nyckelord: MELAS syndrom, epilepsi, klinisk bild, diagnostik, behandling.
MELAS syndrom är en genetiskt betingad sjukdom från gruppen mitokondriella sjukdomar, definierad som mitokondriell encefalomyopati med laktacidos och strokeliknande episoder (mitokondriell encefalomyopati, laktacidos med strokeliknande episoder).
MELAS-syndromet identifierades först som en oberoende nosologisk form av S. Pavlakis et al. år 1984. Ett antal författare föreslår dock att sjukdomen beskrevs tidigare under namnet "familj polyodystrofi, mitokondriell myopati, mjölksyraemi."
Prevalensen i befolkningen har inte fastställts. År 2000 publicerades mer än 120 observationer av MELAS-syndromet, inklusive i inhemsk press.
MELAS-syndromet är i 25 % av fallen maternellt ärftligt med hög risk, men hos 56-75 % av patienterna är familjehistorien inte belastad. Sjukdomen är associerad med mutationer i mitokondriella DNA-gener som kodar för subenheter av respiratoriska kedjekomplex och transport-RNA-gener (MT-ND1, MT-ND5, MT-TH, MT-TL1 och MT-TV). I 80-90 % av fallen av MELAS-syndrom är sjukdomen baserad på en punktmutation i MT-TL1-genen som kodar för leucinöverförings-RNA. Med denna mutation ersätts adeninukleotiden av guanin i position 3243 (A3243G), vilket stör syntesen av alla proteiner i mitokondrier.
Alla organ och vävnader är involverade i den patologiska processen, men muskel- och nervsystemet lider i större utsträckning.
Mukhin K.Yu., Mironov M.B., Nikiforova N.V., Mikhailova C.V., Chadaev V.A., Alikhanov A.A., Ryzhkov BN., Petrukhin A.S.
Epilepsi i MELAS Syndrome Rus. zhur. det. Neur.: volym IV, nr. 3, 2009.
ORIGINALARTIKLAR
ämnen som de mest flyktiga. Svårighetsgraden av kliniska manifestationer beror på tröskeleffekten (ålder, energibehov hos vävnader), på kontrollen av kärngener över syntesen av andningskedjan, heteroplasmi (olika innehåll av muterade mtDNA-molekyler i vävnader). Det har visat sig att hos patienter med MELAS-syndromet är innehållet av mutant mtDNA i olika vävnader 93-96%. Hos probandfamiljemedlemmar detekteras mutant mtDNA också i vävnaderna, men dess innehåll är betydligt lägre: 62-89% i den raderade formen av sjukdomen, från 28 till 89% i frånvaro av kliniska tecken på syndromet.
Sjukdomen utvecklas oftast i en ålder av 6 till 10 år, men det finns fall av en tidigare (upp till två år) eller senare debut - från 21 till 40 år. Innan sjukdomen börjar utvecklas 90-100% av patienterna normalt. Sjukdomsförloppet är progressivt, mer elakartat med en tidig debut.
I de flesta fall manifesterar sjukdomen sig med epileptiska anfall, återkommande huvudvärk, kräkningar och anorexi. Du bör också vara uppmärksam på intolerans mot fysisk aktivitet i form av försämring av hälsan och uppkomsten av muskelsvaghet. Det myopatiska symtomkomplexet manifesteras av träningsintolerans, muskelsvaghet, trötthet och ibland muskelhypotrofi.
När sjukdomen fortskrider utvecklas vanligtvis demens. Symtom som cerebellär ataxi, neurosensorisk dövhet och perifer polyneuropati är mindre vanliga.
Strokeliknande episoder är karakteristiska, som kan manifesteras av återkommande attacker av huvudvärk, yrsel, utveckling av fokala neurologiska symtom (pares, hemianopsi) och koma. Dessa akuta episoder utlöses ofta av feber eller interkurrenta infektioner. Dessa manifestationer kan ha en ganska snabb regression (från flera timmar till flera veckor), såväl som en tendens till återfall.
Epilepsi är en viktig klinisk manifestation som ofta förekommer i de tidiga stadierna av MELAS. Detta är
ofta den mest uppenbara neurologiska manifestationen, särskilt vid atypisk mitokondriell encefalopati (ME). Epileptiska anfall är det första igenkännbara symtomet vid mitokondriell encefalopati (ME) i 53 % av fallen.
Vid MELAS är occipital epilepsi (SE) vanligast. Kännetecknas av fokala anfall med ursprung i de occipitalloberna. Anfall är ofta förknippade med övergående eller ihållande neurologiska symtom som synfältsförlust.
Anfall som härrör från den occipitala cortex är uppdelade enligt deras manifestationer i subjektiva förnimmelser (aura) och kliniskt detekterbara symtom, som regel, med en motorisk komponent. Epileptiska auror som kommer från nackloben inkluderar enkla och komplexa visuella hallucinationer, amauros. De mest typiska anfallen som är karakteristiska för SE är enkla synhallucinationer, som kan visa sig som positiva (blixtar, ljusfläckar) och negativa symtom (skotom, hemianopsi). Oftast beskrivs visuella hallucinationer som en punkt eller fläckar av ljus, antingen stadigt eller blinkande. Som regel är fläcken vit med en grönaktig nyans. Hallucinationer kan också vara flerfärgade eller monokromatiska. Hallucinationer uppträder vanligtvis i synfälten kontralateralt till excitationsfokus i occipital cortex med efterföljande spridning. Det bör dock noteras att i klagomål från patienter upptäcks inte den visuella auran ofta.
Komplexa visuella hallucinationer noteras när epileptisk excitation sprider sig till de occipito-temporala eller occipito-parietala regionerna. Komplexa visuella hallucinationer kan uppträda i form av människor, djurföremål eller scener, vara bekanta eller obekanta, trevliga eller skrämmande, skrämmande, enkla eller groteska, kan vara statiska eller röra sig i ett horisontellt plan och försvinna. Som regel är de ett terminalt symptom före utvecklingen av en motorisk attack; kan vara det första iktala symtomet, men uppstår oftare efter det
VOLYM IV NUMMER 3 2009
grundläggande hallucinationer.
Ictal ama vrosis är en speciell, extremt svårdiagnostiserad typ av anfall som kommer från occipital cortex. Enligt många författare är detta samma vanliga symptom på irritation av occipitalloben, såväl som visuella hallucinationer, men förblir ofta okända. Vanligtvis skiljer patienter inte detta symptom separat i attackens struktur. Synförlust uppstår bilateralt med förlust av sidofält. Möjlig homonym hemianopi kontralateralt till attackens fokus. Patienternas förnimmelser beskrivs av dem som mörkare i ögonen, "vitt mörker", nedsatt färguppfattning. Kanske ett statusförlopp med bildandet av den så kallade status epilepticus amauroticus.
Occipitala anfall kan ge autonoma symtom. Dessa inkluderar migränhuvudvärk, yrsel, illamående och kräkningar. Ett vanligt symptom är migränliknande huvudvärk efter attacken.
De kliniska manifestationerna av anfall som förekommer begränsat i occipital cortex kännetecknas av avvikelse av ögonen åt sidan. Ögonens avvikelse kan noteras tillsammans med huvudets avvikelse åt sidan. I de flesta fall noteras avvikelse av ögonen mot kontralateralt fokus. Däremot beskrivs fall när bortförande av ögonen observeras mot fokus. En av egenskaperna hos "occipital" anfall är också den omedelbara fördelningen av urladdningen till de främre delarna av hjärnan, medan den kliniska bilden som regel domineras av en uttalad motorisk komponent. Toniska, tonisk-kloniska (både hemikonvulsiva och sekundära generaliserade), automotoriska anfall är möjliga. I detta avseende är det viktigt att identifiera de initiala kliniska symptomen - ett omotiverat och plötsligt stopp av blicken, titta på icke-existerande föremål, ett orimligt leende, vegetativa manifestationer och nödvändigtvis dokumentera den primära iktogena zonen med VEM-metoden.
Med utvecklingen av sjukdomen noteras epilepsiresistens mot terapi, ofta med ett statusförlopp. Fall av transformation till Kozhevnikov epilepsi beskrivs. Ett antal auto-
Rov beskriver möjligheten av status epilepticus som det första symtomet hos patienter med MELAS utan tidigare epileptiska anfall. Ribacoba R. et al. beskriver i sin publikation 4 fall av utveckling av epilepsia partialis continua med fokala motoriska anfall, som föregicks av en historia av episoder av migränhuvudvärk. Miyazaki M. et al. visade möjligheten till fortsatt fokal myoklonus inom epilepsia partialis continua hos patienter med MELAS. Araki T. et al. observerade en patient vid en ålder av 37 år med epileptisk status av fokala anfall i form av fluktuationer i medvetandet, homonym hemianopsi i kombination med paroxysmala episoder av ögonavvikelse åt sidan. EEG registrerade fortsatta EEG-mönster av anfall lokaliserade i den occipitala regionen. Hos vuxna patienter med MELAS är det en övervikt av fokala motoriska anfall, men EEG visar en övervikt av multiregional epileptiform aktivitet i de occipitala regionerna.
Epileptiform aktivitet registreras i 71 % av fallen efter anfallens början. En elektroencefalografisk studie av patienter med MELAS-syndrom kännetecknas av epileptiform aktivitet i de occipitala regionerna. Ett antal författare associerar uppkomsten av regionala epileptiforma störningar med stroke. Enligt Fujimoto S:s studie, under den akuta perioden (dvs inom 5 dagar efter en strokeliknande episod), hade majoriteten av undersökta patienter med MELAS-syndrom regionala högamplitud deltavågor i kombination med polyspikar. Författarna föreslår att man ska betrakta detta mönster som patognomoniskt för strokeliknande episoder. Förutom de occipitala regionerna kan epileptiform aktivitet spridas till de temporala regionerna, bifrontalt och även bilateralt till de bakre regionerna med diffus distribution. Kanske uppkomsten av ett fotoparoxysmalt svar under rytmisk fotostimulering.
Det ledande laboratorietecknet är en ökning av nivån av laktat i blodet.
ORIGINALARTIKLAR
wi över 2,0 mmol / l, vilket leder till utvecklingen av laktacidos.
En MR av hjärnan i de tidiga stadierna av sjukdomen kan vara omärklig, även om epilepsi inträffar. Neuroimagingmetoder avslöjar infarktzoner i hjärnhalvorna (80 %), mindre ofta i lillhjärnan och basala ganglierna. Det kan också förekomma förkalkning av basalganglierna, atrofi av hjärnbarken. I en fotonemissionsstudie detekteras ackumuleringen av isotopen 3-16 dagar före uppkomsten av infarktzonen (minskning av isotopsignalen) på ett datortomogram av hjärnan. MRT av hjärnan visar lesioner främst lokaliserade i de occipitalloberna, vilka kan vara övergående. Occipital cortex är övervägande påverkad, den vita substansen skadas i mindre utsträckning. På T2-viktade bilder uppträder hjärnskador i MELA som områden med ökad signalintensitet. Övergående hyperintensiva områden associeras av ett antal författare med reversibelt kärlödem.
Angiografi avslöjar vanligtvis inte vaskulära abnormiteter. Diffusionsviktad MRT visar förändringar associerade med vasogent ödem.
Histopatologi: Muskelbiopsi avslöjar fibrer med trasiga "röda kanter". Obduktion av hjärnan kännetecknas av en kombination av gamla och nya foci av infarkter, såväl som atrofi av cortex med fokala foci av nekros.
För närvarande är terapi stödjande. Behandlingens huvudsakliga inriktning är att förbättra energibalansen i mitokondrierna och andningskedjan. Applicera koenzym p10 (80-300 mg / dag), vitaminer K1 och KZ (25 mg / dag), bärnstenssyra (upp till 6 g / dag), vitamin C (2-4 g / dag), riboflavin (100 mg / dag) dag) och nikotinamid (upp till 1 g/dag). I samband med att den sekundära karnitinbristen utvecklas, ordineras patienterna L-karnitin (upp till 100 mg/kg/dag). Vitamin E (300-500 mg/dag) och vitamin C (2-4 mg/dag) används som antioxidantterapi.
Det finns inga allmänt accepterade antiepileptiska behandlingsregimer för MELA. Ett antal författare föreslår att man utesluter läkemedel som kan hämma energiomsättningen (barbiturater, valproinsyraläkemedel; såväl som vissa läkemedel från andra grupper, till exempel kloramfenikol). Litteraturen beskriver flera isolerade fall av anfallsförvärring med användning av valproinsyra vid MELA-syndromet med A3243C-mutationen. De huvudsakliga AED:erna vid behandling av epilepsi vid MELA-syndromet anses vara tegretol (eller trileptal), topamax, keppra i genomsnittliga terapeutiska doser. Korrekt vald terapi leder till en signifikant minskning av frekvensen av sekundära generaliserade konvulsiva anfall. Anfall med nedsatt vegetativ-visceral och visuell funktion är dock vanligtvis resistenta mot behandling. I det terminala stadiet av sjukdomen kan frekvensen av epileptiska anfall minska.
Här är fallhistorien för en patient med diagnosen MELAY syndrom verifierad under sin livstid.
Patient Ch.A., 11 år, observerades vid Center for Pediatric Neurology and Epilepsy. Vid intagningen framfördes klagomål på en gradvis förlust av talförmåga, en uttalad gångstörning med vägran att gå, en signifikant minskning av synen, nyckfullhet och negativt beteende; dagliga serieangrepp i form av ryckningar i ansiktsmusklerna, musklerna i de övre och nedre extremiteterna, samt kortvariga episoder av synförlust.
Sjukdomens debut noterades vid en ålder av 5 år 9 månader. För första gången, mot bakgrund av fullständig hälsa, när man somnade, uppträdde en svår huvudvärk, enkla visuella hallucinationer ("gul stråle"), följt av en våldsam vändning av ögonen och huvudet åt sidan och utvecklingen av en generaliserad tonisk-kloniskt krampanfall, varefter kräkningar noterades. Efter 9 månader attacker med samma symtom återkom och fick snabbt en seriekaraktär. Efter utnämningen av tegretol i en dos på 400 mg per dag, minskade frekvensen av attacker till 1 gång per månad. Tegretol ersattes av Depakine Chrono i en dos på 900 mg/dag, mot vilken en klinisk remission noterades under 6 månader. Med tanke på det kliniska symtomet
VOLYM IV NUMMER 3 2009
tomatik, inskränkning av anfall till insomningsperioden, normal intelligens hos patienten, en positiv reaktion på valproat, idiopatisk occipital epilepsi diagnostiserades.
Vid 7 års ålder återupptogs fokala versiva anfall med sekundär generalisering vid insomning med samma frekvens 1 gång per månad. En ökning av dosen av Depakine till 1500 mg/dag ledde inte till en minskning av frekvensen av anfall. När lamiktal tillsattes i en dos av 75 mg/dag upphörde attackerna i 4 månader och återupptogs sedan med samma frekvens. Vid 8 års ålder anslöt sig attacker med kortvarig synförlust. Från 8 år 8 månader innan man somnade började atypiska frånvaron dyka upp: snabb blinkning med stängning av ögonlocken och inrättandet av ögongloberna uppåt; medvetandet fluktuerar.
Vid 9 års ålder uppträdde flera serieangrepp, som varade i flera dagar, med enkla synhallucinationer i form av en blinkande "stråle" framför ögonen, med en vridning av ögonen och huvudet åt höger. Innan man somnade förvandlades sådana attacker ibland till fokala hemikloniska, som manifesterades genom minskning av ansiktsbehandlingen
muskulatur till höger, ryckningar i huvudet till höger, klonier i höger extremiteter (större än armen). Ibland efter attacken var det kraftig huvudvärk och kräkningar. I samma ålder uppträdde hämmande attacker: en aura i form av gåshud i stortån på höger fot, följt av kortvarig svaghet i höger ben och obekvämhet i höger hand. Topamax introducerades i behandlingsregimen med en dos på 100 mg/dag - det förekom inga epileptiska anfall under 1 år.
Vid 9 års ålder uppträdde också paroxysmala tillstånd, åtföljda av svår huvudvärk, kräkningar och utveckling av högersidig hemipares. I vissa fall åtföljdes sådana tillstånd av amauros som varade från flera minuter till flera dagar.
Vid 10,5 års ålder återkom attacker i form av vridning av huvudet till vänster, ryckiga rörelser av ögongloberna till vänster, som varade upp till 5 s, frekvens upp till 3 gånger per timme, dagligen, även under sömnen. Topamaxdosen ökades till 150 mg/dag utan signifikant effekt. Vid 10 år 10 månader. efter en intensiv huvudvärk, omväxlande
Ris. 1. Patient Ch.A. 10 år. Diagnos: MEAE-syndrom. Symtomatisk fokal epilepsi.
Video-EEG-övervakning (2004): mot bakgrund av en diffus nedgång i hjärnans huvudaktivitet registrerades fortsatt epileptiform aktivitet i den vänstra occipitalregionen. Subkliniska EEG-mönster av en attack registrerades också i den vänstra occipitalregionen med spridning till den vänstra posteriora temporala regionen.
Centrum för pediatrisk neurologi och epilepsi
under ledning av professor K.Yu. Mukhina är engagerad i diagnostik och behandling av ångestsjukdomar i nervsystemet i Aetei, specialiserat på etiska former av epilepsi.
Huvudriktningar
aktiviteter:
Epilepsi hos barn och ungdomar
Huvudvärk
Sömnstörningar hos barn
Tiki, enures
Undersökning av barn under de första ^ månaderna av livet.
Tentamen i vårt center:
Diagnos och behandling av sjukdomar i nervsystemet hos barn
Fullständig diagnostik (inklusive förkirurgi) och behandling av epilepsi
Konsultation av neurologer och epileptologer
Konsultation av en barnläkare (ofta sjuka barn, gastroenterologi, etc.)
Konsultation av psykiater och psykolog.
Genetisk konsultation med tester (inklusive karyotyping)
Video-EEG-övervakning (i specialutrustade rum på centret eller med ett besök i patientens hem)
Dator (digital) elektroencefalografi
UZDG (ultrasounddopplerografi) av kärlen i huvudet och nacken
Echoencefalografi (ECHO EG)
På vår sida kan du prenumerera på tidskriften "Russian Journal of Child Neurology" via Internet.
Detaljerad information om centrets arbete från 10:00 till 19:00 per telefon:
Tel.: (+7495) 983-09-03; (+7926)290-50-30 Tel./Fax: (+7495) 394-82-52
Adress: st. Borisovskie Prudy, 13, byggnad. 2. Internet: www.epileptologist.ru E-post: [e-postskyddad](för en detaljerad ruttkarta, se hemsidan)
VOLYM IV NUMMER 3 2009
fokala hemkloniska och sekundära generaliserade anfall som blev seriella och varade i 48 timmar. Frizium tillsattes topamax i en dos på 10 mg/dag med en tillfällig positiv effekt.
Från 8 års ålder började svårigheter med assimileringen av skolmaterial att noteras; minskat minne. Det var ökad trötthet, utmattning, hämning av mental aktivitet. Pojken blev nyckfull, irriterad, negativ; bakgrunden till humöret har minskat. Från 9 års ålder skedde en ökning av denna symptomatologi.
Från livets anamnes är det känt att barnet föddes från en andra normal graviditet, en andra termin, födelsevikt 2800 g, längd 53 cm. Tidig psykomotorisk utveckling och talutveckling var helt förenlig med åldern. Tidigare sjukdomar: vattkoppor vid 6 års ålder, frekventa akuta luftvägsvirusinfektioner (upp till 4 gånger per år) från 6 år. Ärftlighet för epilepsi och andra neurologiska sjukdomar är inte belastad.
Vid undersökningstillfället (11 år) var barnets tillstånd allvarligt; reagerar negativt på besiktning. Medveten, pro-orienterad
rum och tid. Han kommer extremt motvilligt i kontakt, vägrar följa instruktioner. Spontan nystagmus till vänster, huvudet lutat till vänster axel med en vridning åt höger. Tungan är i mittlinjen, svalgreflexen är reducerad; Dysfagi och dysartri noteras. Synen är nedsatt.
Måttlig diffus muskulär hypotoni bestäms. Senreflexerna reduceras jämnt. Det var en liten minskning av muskelstyrkan i de högra extremiteterna. Patologiska fotreflexer upptäcktes inte. Det finns inga objektiva uppgifter för brott mot känslighet. Inte värt det i Rombergtestet. vägrar gå. När du försöker sätta honom på fötterna gråter han, sätter sig på golvet. Saknas vid utförande av ett finger-index-test. Talar långsamt, med enstaka ord, motvilligt.
Ytterligare undersökningsmetoder. Video-EEG-övervakning (2004). Betydande avmattning av den huvudsakliga bakgrundsinspelningsaktiviteten. Under studien registrerades fortsatt epileptiform aktivitet i den vänstra occipitalregionen med spridning till den vänstra posteriora temporala regionen och med periodisk bildning av ett EEG-mönster
född 1993 16/12/05
Ris. 2. Patient Ch.A. 11 år. Diagnos: MELAS syndrom. Symtomatisk fokal epilepsi.
Video-EEG-övervakning utfördes i dynamik efter 1 år (2005): en betydande nedgång i hjärnans bakgrundsaktivitet. Under sömnregistrering registreras fortsatt regional deceleration i den högra fronto-centrala regionen, i vars struktur toppvågsaktivitet detekteras i den högra fronto-centrala regionen.
ORIGINALARTIKLAR
stupa (fig. 1). Även fortsatt regional retardation i den högra fronto-centrala regionen med inkluderandet av enstaka skarpa vågor bestäms.
Video-EEG monitoring in dynamics (2005): Betydande nedgång i hjärnans bakgrundsaktivitet. Studien registrerade fortsatt regional nedgång i den högra fronto-centrala regionen. I strukturen för regional retardation i det högra fronto-centrala området avslöjas toppvågsaktivitet (Fig. 2).
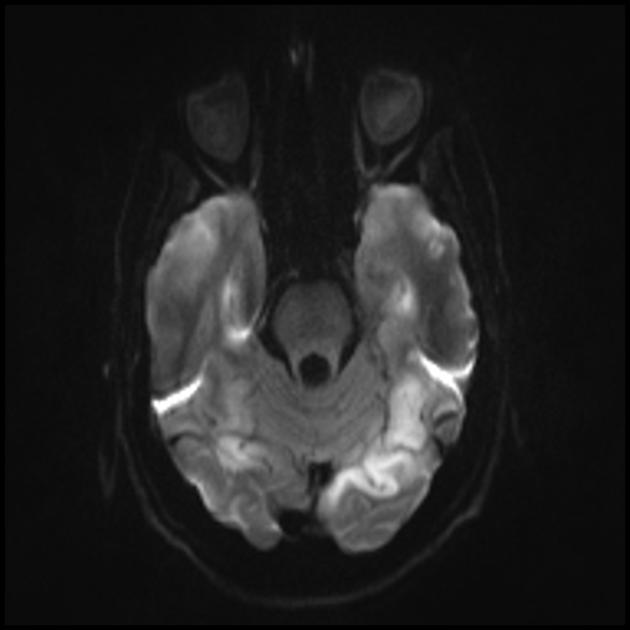
MRT av hjärnan. Den första magnetröntgen (6 år) avslöjade en enda hyperintensiv signal i T2-läge i den vänstra hjärnhalvan av lillhjärnan. MRT-studie över tid (10,5 år): en signifikant försämring av den primära lesionen avslöjades med spridningen av den patologiska processen brett till vänster och höger occipital-parietalregioner i båda hjärnhalvorna (Professor A.A. Alikhanov).
Visuellt framkallade potentialer: signifikanta morfologiska och funktionella förändringar i det visuella afferenta systemet i nivå med synnerven och den kortikala delen av den visuella analysatorn, mer uttalad till vänster.
Ögonläkares konsultation: partiell atrofi av synnerverna. Element av kortikal agnosi.
Elektrokardiogram: ektopisk rytm med acceleration upp till 100 slag per minut.
Vertikal position för hjärtats elektriska axel. Förändringar i repolarisationsprocesser, som är mer uttalade vid ortostas.
Elektroneuromyografi: avslöjade den primära muskeltypen av lesion. Ledningshastigheterna längs de perifera nerverna reduceras inte.
Studien av nivån av laktat i blodet: innehållet av laktat i blodet är 3,0 mmol / l (normen är upp till 1,8).
Med hänsyn till förekomsten av epileptiska anfall som kommer från de occipitala regionerna i hjärnbarken, resistenta mot terapi, strokeliknande episoder, perioder av amauros, kognitiv försämring, närvaron av hyperintensiva signaler i cerebellum och bakre regioner av hjärnbarken på MRT, en ökning av nivån av laktat i blodet, patienten hade en diagnos av MELAS syndrom föreslogs. Under en genetisk undersökning hittades A3243G-mutationen i det heteroplasmatiska tillståndet i blodkropparna (diagnosen utfördes vid State Research Center vid den ryska akademin för medicinska vetenskaper), och diagnosen verifierades.
Observation vid uppföljning visade en snabb progression av kränkningar av högre mentala funktioner, utveckling av kortikal blindhet, fullständig orörlighet hos patienten, följt av början av döden vid en ålder av 12 år 10 månader. (efter 7 år från början av sjukdomen).
Bibliografi
1. Nikolaeva E.A., Temin P.A. Mitokondriella sjukdomar åtföljda av nedsatt neuropsykisk utveckling. MELAS syndrom // Ärftliga störningar i den neuropsykiska utvecklingen hos barn. En guide för läkare redigerad av Temin P.A. Kazantseva L.Z. - Medicin, 2001. - S. 96-107.
2. Nikolaeva E.A., Temin P.A., Nikanorova M.Yu., Klembovsky A.I., Sukhorukov V.S., Dorofeeva M.Yu., Korsunsky A.A. Behandling av ett barn med mitokondriellt syndrom MELAS (mitokondriell encefalopati, laktacidos, strokeliknande episoder) // Russian Bulletin of Perinatology and Pediatrics. - 1997. - Nr 2. - S. 30-34.
3. Smirnova I.N., Kistenev B.A., Krotenkova M.V., Suslina ZA. Strokeliknande förlopp av mitokondriell encefalomyopati (MELAS syndrom) // Atmosfera. Nervsjukdomar. - 2006. - Nr 1. - S. 43-48.
4. Temin PA, Nikanorova M.Yu., Nikolaeva E.A. MELAS-syndrom (mitokondriell encefalomyopati, laktacidos, strokeliknande episoder): huvudsakliga manifestationer, diagnostiska kriterier, behandlingsalternativ // Nevrol. tidskrift - 1998. - Nr 2. - S. 43-48.
5. Ajmone-Marsan C., Ralston B. Det epileptiska anfallet, dess funktionella morfologi och diagnostiska betydelse. - Springfield (IL): Charles C. Thomas, 1957. - S. 3-231.
6. Aldrich M.S., Vanderzant C.W., Alessi A.G., Abou-Khalil B., Sackellares J.C. Ictal kortikal blindhet med permanent synförlust // Epilepsi. - 1989. - V. 30. - S. 116-20.
7. Araki T., Suzuki J., Taniwaki Y., Ishido K., Kamikaseda K., Turuta Y., Yamada T. Ett fall av MELAS som presenterar komplex partiell status epilepticus // Rinsho Shinkeigaku. - 2001. - V. 41(8). - S. 487-90.
VOLYM IV NUMMER 3 2009
8. Canafoglia L., Franceschetti S., Antozzi C., Carrara F., Farina L., Granata T., Lamantea E., Savoiardo M., Uziel G., Villani F., Zeviani M., Avanzini G. Epileptic fenotyper associerade med mitokondriella störningar // Neurologi. - 2001. - V. 56(10). - S. 1340-6.
9. Chih-Ming Lin, Peterus Thajeb. Valproinsyra förvärrar epilepsi på grund av MELAS hos en patient med en A3243G-mutation av mitokondriellt DNA // Metab Brain Dis. - 2007 - V. 22(1). - S. 105-109.
10. Chinnery P.F., Howell N., Lightowlers R.N. et al. Molekylär patologi hos MELAS och MERRF. Sambandet mellan mutationsbelastning och kliniska fenotyper // Hjärna. - 1997. - V.120. - P. 1713-1721.
11. Durand-Dubief F., Ryvlin P, Mauguiere F. Polymorfism av epilepsi associerad med A3243G-mutationen av mitokondriellt DNA (MELAS): orsaker till försenad diagnos // Rev Neurol (Paris). - 2004. - V. 160(8-9). - s. 824-829.
12. Dvorkin G., Andermann F., Carpenter S. Klassisk migrän, svårbehandlad epilepsi och flera stroke: ett syndrom relaterat till mitokondriell encefalopati / I: Andermann F., Lugaresi E., redaktörer. migrän och epilepsi. - Boston: Butterworths, 1987. - S. 203-32.
13. Fujimoto S., Mizuno K., Shibata H., Kanayama M., Kobayashi M., Sugiyama N., Ban K., Ishikawa T., Itoh T., Togari H., Wada Y. Serial electroencephalographs-fynd hos patienter med MELAS // Pediatr Neurol. - 1999. - V. 20(1). - S. 43-48.
14. Goto Y., Nonaka I., Horai S.A. En mutation i tRNA leu(UUR)-genen associerad med MELAS-undergruppen av mitokondriella encefalomyopatier // Nature. - 1990. - V. 348. - P. 651-653.
15. Hasuo K., Tamura S., Yasumori K., Uchino A., Goda S., Ishimoto S., et al. Datortomografi och angiografi vid MELAS (mitokondriell myopati, encefalopati, laktacidos och strokeliknande episoder): rapport om 3 fall // Neuroradiology. - 1987.-V. 29. - s. 393-397.
16. Hirano M., Pavlakis S.G. Mitokondriell myopati, encefalopati, laktacidos och strokeliknande episoder (MELAS): Aktuella begrepp // J. clin. Neurol. - 1994. - V. 9. - P. 4-13.
17. Hori A., Yoshioka A., Kataoka S., Furui K., Tsukada K., Kosoegawa H., Sugianto, Hirose G. Epileptiska anfall hos en patient med mitokondriell myopati, encefalopati, mjölksyra och strokeliknande episoder ( MELAS) // Jpn J Psychiatry Neurol. - 1989. - V. 43(3). - s. 536-537.
18. Kuriyama M., Umezaki H., Fukuda Y., Osame M., Koike K., Tateishi J., et al. Mitokondriell encefalomyopati med laktat-pyruvatförhöjning och hjärninfarkter // Neurologi. - 1984. - V. 34. - P. 72-77.
19. Kuzniecky R. Symtomatisk occipitallobsepilepsi // Epilepsi. - 1998. - V. 39 Suppl 4. - S. 24-31.
20. Ludwig B.I., Ajmone-Marsan C., Van Buren J. Djup och direkt kortikal registrering av anfallsstörningar av extratemporalt ursprung // Neurology. - 1976. - V. 26. - P. 1085-1099.
21. Ludwig B.I., Ajmone-Marsan C. Clinical ictal patterns in epileptic patients with occipital electroencephalo-graphic foci // Neurology. - 1975. - V. 25. - P. 463-471.
22. Matthews P.M., Tampieri D., Berkovic S.F., Andermann F., Silver K., Chityat D., et al. Magnetisk resonanstomografi visar specifika avvikelser i MELAS-syndromet // Neurologi. - 1991. - V. 41. - P. 1043-1046.
23. Miyazaki M., Saijo T., Mori K., Tayama M., Naito E., Hashimoto T., Kuroda Y., Nonaka I. Ett fall med MELAS associerat med epilepsia partialis continua // No To Hattatsu. - 1991. - V. 23(1). - S. 65-70.
24. Montagna P., Gallassi R., Medori R., Govoni E., Zeviani M., Di Mauro S., et al. MELAS-syndrom: karakteristiska migrän- och epileptiska egenskaper och maternell överföring // Neurologi. - 1988. - V. 38. - P. 751-754.
25. Ooiwa Y., Uematsu Y., Terada T., Nakai K., Itakura T., Komai N., et al. Cerebralt blodflöde vid mitokondriell myopati, encefalopati, laktacidos och strokeliknande episoder // Stroke. - 1993. - V. 24. - P. 304-309.
26. Pavlakis S.G., Phillips P.C., Di Mauro S. et al. Mitokondriell myopati, encefalopati, laktacidos och strokeliknande episoder: Ett distinkt kliniskt syndrom // En neurol. - 1984. - V. 16. - P. 481-488.
27. Ribacoba R., Salas-Puig J., Gonzalez C., Astudillo A. Karakteristika för status epilepticus i MELAS. Analys av fyra fall // Neurologia. - 2006. - V. 21(1). - S. 1-11.
28. Williamson P.D., Spencer S.S. Kliniska och EEG-egenskaper hos komplexa partiella anfall av extratemporalt ursprung // Epilepsi. - 1986. - V. 27 (Suppl 2). - S. 46-63.
29. Williamson P.D., Thadani V.M., Darcey T.M., Spencer D.D., Spencer S.S., Mattson R.H. Occipitallobsepilepsi: kliniska egenskaper, spridningsmönster för anfall och resultat av operation // Ann Neurol. - 1992. - V. 31. - S. 3-13.
30. Yi-Min Chen, Chih-Ming Lin, Peterus Thajeb. Paradoxal effekt av natriumvalproat som förvärrar epilepsi av MELAS hos en patient med A3243G-mutation av mitokondriella DNA // Central European Journal of Medicine. - 2007. - V. 2(1). - S.103-107.
31. Yoneda M., Maeda M., Kimura H., Fujii A., Katayama K., Kuriyama M. Vasogent ödem på MELAS: en seriell studie med diffusionsvägd MR-avbildning // Neurology. - 1999. - V. 53. - P. 2182-2184.
MELAS syndrom (MELAS) beskrevs första gången 1984 av S. Pavlakis och kollegor. Men vissa forskare tror att syndromet redan tidigare betecknades av sådana begrepp som familjär polyodystrofi, mjölksyraemi.
Kärnan i patologi
År 1994 beskrev S. Pavlakis och Mizio Hirano 110 fall av sjukdomen. MELAS (mitokondriell encefalomyopati, laktacidos och strokeliknande episoder) är en multisystem progressiv neurodegenerativ sjukdom. Den är polymorf och kännetecknas av encefalopati med kramper och demens, laktacidos. Sjukdomen orsakas av mutationer i mitokondrie-DNA (mtDNA). Denna sjukdom har ett annat namn - mitokondriell encefalomyopati.
Allmän information
Från 25 till 44% av fallen av sjukdomen är ärftliga, överförs genom moderlinjen. I andra fall inträffar det för första gången. För närvarande är mer än 10 gener kända som muterar och leder till utvecklingen av detta syndrom. Dessa är gener som kodar för överförings-RNA:s funktioner. MELAS-syndrom avser mitokondriella sjukdomar (MD) med en onormal ansamling av mitokondrier, vilket resulterar i störningar av hela cellens energimetabolismsystem.
Sjukdomar i denna grupp överförs endast genom moderlinjen. Med dem påverkas de mest energiberoende organen och vävnaderna i olika kombinationer: skelettmuskler, hjärta, hjärna, syn, lever och njurar.
Symtomen är polymorfa och kan uppträda i alla åldrar. Det inkluderar manifestationer av diabetes, hörselnedsättning, kramper, endokrinopatier, kortväxthet, hjärtpatologier, absolut oförmåga att träna och psykomotoriska avvikelser.
Innan det är den psykomotoriska utvecklingen helt normal. Inga patienter med samma symtom identifierades, eftersom mutationen påverkar många gener: MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTTS2, MTND1, 5, 6. Deras antal fortsätter att öka.
Hos 80 % av patienterna orsakas MELAS-syndromet av en punktsubstitution A3243G i leucin-tRNA-genen (UUR).
Frekvensen har studerats opålitligt. Det finns bara ett fåtal data: till exempel i Finland var A3243G-mutationshastigheten 16:100 tusen av befolkningen; i England - 1 fall per 13 tusen personer.
Patologiska förändringar

Den karakteristiska patologiska egenskapen för MELAS syndrom är trasiga röda fibrer (RRF), som kan ses i muskelvävnad med en speciell Gomory tricolor. De är resultatet av muterade gener och det morfologiska substratet av mtDNA-skada, som bildas som ett resultat av spridningen av dessa onormala mitokondrier.
Vad är en mitokondrie förresten
Mitokondrier är en tvåmembransorganell i en eukaryot cell (en cell som har en kärna), vars huvudsakliga funktion är att tillföra energi. Det vill säga, faktiskt, mitokondrier är energibasen för celler, deras energistationer.
Antalet mitokondrier i celler kan variera under hela dess liv från några till tusentals. Och fler av dem händer i celler associerade med energiproduktion.
Mitokondrierna själva är oftast runda långsträckta, i storlek från 1 till 10 mikron. De kan frysa orörligt eller röra sig inuti cellens cytoplasma. De flyttar vanligtvis dit mer energi behövs.
På mitokondriernas inre membran finns utväxter (cristae) på vilka det finns hela system av enzymer. I grund och botten är dessa proteinföreningar. Antalet cristae beror på intensiteten av syntesprocesser. Till exempel, i muskelcellernas mitokondrier finns det alltid många av dem.
Mitokondrier har ett autonomt proteinsyntessystem - DNA, RNA och ribosomer. Några av de nödvändiga proteinerna syntetiseras av mitokondrierna själva - 5%, och några erhålls från cytoplasman - 95%. Energi utvinns från organiska föreningar genom en mängd olika enzymatiska reaktioner.
Vissa av dessa reaktioner sker med deltagande av syre, det vill säga oxidation sker, och efter andra frigörs CO 2 med överföring av väteprotoner och frigöring av energi. Med andra ord är mitokondriet en aktiv deltagare i cellandningen.
Dessa reaktioner sker på cristae eller i själva mitokondrierna, vilket är så viktigt för cellen att om det botas blir cellen helt frisk.
Patogenes

Vid första anblicken, vid MELAS-syndrom, liknar tillståndet en variant efter stroke. Men i själva verket är det atypiskt: det förekommer hos unga människor, ofta provocerade av infektionssjukdomar, och kan uppstå i form av en elakartad migränliknande huvudvärk, kramper.
Angiografi ger inga vaskulära patologier. Det kan finnas normala kärl eller ökade kaliber av vissa artärer, vener eller kapillär hyperemi inträffar.
MRT visar att akuta hjärnskador vid MELAS-syndrom kan migrera och till och med försvinna. Vissa foci fluktuerar. För en typisk stroke är detta helt okarakteristiskt.
Vid MELAS-syndromet förekommer multifokal nekros. För det mesta märks detta i den occipitala delen (posterior lokalisering) av hjärnbarken och den vita substansen i subcortex. Men de kan också förekomma i andra delar av hjärnan. Dessa områden liknar nekros vid en hjärtinfarkt, men är belägna utanför bassängerna i de centrala hjärnkärlen.
Symtomatiska manifestationer

Vanligtvis debuterar MELAS-syndrom hos barn vid 6-10 års ålder (det kan börja vid 3 år och vid 40 år). Tidig debut av sjukdomen är mer typisk och drabbade 90 % av patienterna. Med en tidig debut, flyter sjukdomen svårare. Patienterna är vanligtvis underdimensionerade, muskelsvaga och absolut inte anpassade till fysisk ansträngning.
All spänning eller fysisk aktivitet får dig att må sämre. Av de inre organen påverkas hjärtat av undernäring av muskeln och ledning, följt av utveckling av kardiovaskulär insufficiens. Nefropati, diabetes, gastrointestinala störningar med kräkningar förekommer också, hörseln minskar. Kännetecknas av muskelvärk, brist på reflexer, pareser, kramper, IPE, medvetslöshet. Muskelsvaghet (myopatiskt syndrom) och sensorineural hörselnedsättning är också typiska för denna patologi.
Endokrinopati representeras inte bara av diabetes mellitus, utan också av brist på tillväxthormon. Hjärt- och njursjukdomar är sällsynta vid utvecklingen av sjukdomen i fråga.
Kramper vid MELAS syndrom är mycket varierande. De kan vara fokala, generaliserade, tonisk-kloniska och myokloniska. Absolut okänslighet hos anfall för antikonvulsiv terapi är karakteristisk. Det händer ofta att läkare ställer diagnosen epilepsi och skriver ut till exempel valproinsyra. Efter det försämras hälsotillståndet kraftigt och kramper ökar, eftersom det trycker ner mitokondrier. Även om demens utvecklas, blir det sällan ett tydligt symptom.
Det är också karakteristiskt för sjukdomen, men det förekommer också i många andra patologier, och därför kan det inte tjäna som grund för diagnos. Endast i kombination med migrän, kramper och/eller strokeliknande fenomen kan uppkomsten av MELAS-syndromet misstänkas. Inte ens så omfattande symtom ger en korrekt diagnos. Förloppet av processen sker på olika sätt.
tecken

Det kliniska kännetecknet för MELAS-syndromet är strokeliknande episoder (IPE), där neurologiska symtom plötsligt uppträder. IPE kännetecknas av asymmetri av lesioner. De kan vara flera.
Selektiviteten för sådan lokalisering ger också vissa fokala symtom:
- hemianopsi (kortikal blindhet);
- hemipares;
- sensorisk afasi (missförstånd av ord);
- acalculia (överträdelser av kontot);
- agraphia (stavningsöverträdelser);
- ataxi (försämrad koordination av frivilliga rörelser);
- förändringar i medvetandet.
Det är inte ovanligt att dessa strokeliknande symtom återkommer var 1 till 3:e månad. Det speciella med akuta episoder i MELAS är att de har en snabb regression, men ofta återkommer, det vill säga som om de passerar spårlöst. Dessutom, hos patienter med denna sjukdom, deponeras förkalkningar i basalganglierna (detta finns på CT).
Strokeliknande episoder utvecklas ofta vid 5-15 års ålder. De blir aldrig resultatet av tromboembolism. Angiopati i MELAS beror på hyperproliferation av samma mitokondrier.
IPE i symtom manifesteras av återkommande attacker av cephalalgi, yrsel, pares, förlamning av armar och ben, kranialnerver. Mannen är helt demoraliserad.
Laktacidos vid MELAS syndrom

Dess främsta boven är ett överskott av mjölksyra i blodet och vävnaderna i nervsystemet. Detta minskar kraftigt surheten i blodet i artärerna. Sådan acidos är en frekvent följeslagare av diabetes mellitus, som finns vid MELAS-syndrom.
På ett tidigt stadium är manifestationer ospecifika. Följande symtom observeras: allmän svaghet, bröstsmärtor, apati, dåsighet. Myalgi efter fysisk ansträngning och intermittent snabb andning utan någon lukt är mycket karakteristiska.
I mellanstadiet ackumuleras mjölksyra och hyperventilationssyndrom (HVS) uppstår. Koldioxid ackumuleras i blodet. Det börjar bildas bullriga andetag - Kussmaul. Trycket sjunker för att kollapsa, oliguri sätter in. Patienten blir rastlös, förvirrad och förlorar sedan medvetandet med den efterföljande utvecklingen av koma - detta är det sista steget. Symtom på laktacidos utvecklas snabbt, nämligen inom några timmar. Sedan kommer döden.
Diagnostiska åtgärder

Som redan noterats, på grund av polymorfismen av symtom och mutationen av ett stort antal gener, är diagnosen MELAS-syndrom svår. Hålls:
- allmänna och biokemiska blodprov;
- muskelbiopsi;
- genetisk studie med en jämförande analys bland sjuka släktingar;
- CT av hjärnan: områden av infarkter oftare i hemisfärerna, mindre ofta i lillhjärnan, basala ganglier;
- en ökning av kalibern av blodkärl (artärer, vener, kapillärer);
- DNA-diagnostik: sök efter karakteristiska punktmutationer i mtDNA.
Terapimetoder
Behandling för MELAS syndrom har inte utvecklats, och det är för närvarande obotligt. Det finns bara försök att bromsa nederlagsprocessen. Behandlingen går i två riktningar: postsyndromisk terapi (epilepsi, diabetes mellitus) och patogenetisk. Det finns dock ingen effektiv patogenetisk terapi idag.
Det finns symtomatiska behandlingar: med hörselnedsättning används hörapparater aktivt, med andningsmuskelsvaghet tillhandahålls andningsterapi. Det noterades att med MELAS-syndromet i patientens blod minskar nivån av L-arginin signifikant under IPE. Därför utförs terapi med argininpreparat och vitaminer. Den positiva effekten av koenzym Q eller idebinon (noben), bärnstenssyrapreparat, vitaminer K 1 och K 3, B 2 , B 3 , E, C studeras; L-karnitin, antioxidanter (mexidol, mildronat), laktacidoskorrigerare (dimefosfon) - alla förbättrar cellens energiomsättning. Vid behandling av anfall är valproater och barbiturater inte föreskrivna, eftersom de trycker ned mitokondrier.
Som ett förebyggande av detta syndrom är det bäst att tillgripa IVF-metoden. Om en kvinna vet att hon har ett fall av manifestation av denna sjukdom i sin familj, tas cytoplasman för befruktning från en frisk kvinna. Metoden är fortfarande på studiestadiet, den är inte massa.
Vissa funktioner
Diagnos av mitokondriella störningar kräver en mycket noggrann inställning till terapi. Medel för metabolisk verkan måste nödvändigtvis inkluderas i den. De stabiliserar processerna för vävnadsandning, oxidativ fosforylering i celler. Endast den systematiska implementeringen av sådan behandling kan hjälpa till att upprätthålla patienters tillstånd och förhindra strokeepisoder.
Prognos
Prognosen är ogynnsam på grund av bristen på effektiv behandling. Den förväntade livslängden från första symptomdebut överstiger vanligtvis inte fem år. Bristen på kunskap om orsakerna till sjukdomen leder till att den optimala behandlingsregimen ännu inte har hittats. Allt detta gör att chanserna till bot minimala.
MELAS-syndrom är en mitokondriell störning som kännetecknas av muskel- och CNS-inblandning.
MELAS (eng. Mitokondriell encefalomyopati, laktacidos och strokeliknande episoder - "mitokondriell encefalomyopati, laktacidos, strokeliknande episoder") är en progressiv neurodegenerativ sjukdom som kännetecknas av de manifestationer som anges i titeln, och åtföljs av polymorfa symtom - stroke, diabetes, kramper, minskad hörselnedsättning, hjärtsjukdomar, kortväxthet, endokrinopatier, träningsintolerans och neuropsykiatriska störningar.
Berättelse.
MELAS-syndromet beskrevs första gången 1984 av Pavlakis och kollegor; tio år senare publicerade Pavlakis och Mizio Hirano en recension av 110 fall.
arvstyp:
moderlig
Epidemiologi:
Den exakta frekvensen av sjukdomen är inte känd. Det finns få data i litteraturen om förekomsten av sjukdomen. I norra Finland är A3243G-mutationshastigheten 16,3:100 000.
Patogenes:
Mutationer av mitokondralt DNA, som styr mitokondriernas andningskedja, åtföljs av en störning i processerna för oxidativ fosforylering, den viktigaste energikällan för metaboliska processer i cellen.
Kliniska manifestationer
Vid 40 års ålder tas patienter med MELAS in med en klinik för övergående ischemisk attack, samt med epilepsi, upprepade kräkningar, huvudvärk och muskelsvaghet. Dessa patienter diagnostiseras ofta kliniskt med demens.
Ung ålder och frånvaron av riskfaktorer specifika för stroke gör MELAS mer eftertänksam.
Laboratoriedata
Laktatacidos - ökade nivåer av laktat och pyruvat.
Visualiseringsdata
Förändringar i hjärnan liknar förändringar i en stroke.
Skillnader från en stroke
1) de drabbade områdena sammanfaller inte med gränserna för de arteriella vaskulära poolerna.
2) med upprepade attacker visualiseras foci i en annan lokalisering.
+ kliniska data (ung ålder, inga riskfaktorer för stroke).
CT
Flera hypodensa områden som inte överensstämmer med kärlbädden.
Förkalkning av basalganglierna (vanligast hos äldre patienter).
Atrofi uppstår mot bakgrund av regression och klinisk förbättring.
MRI
Akut infarkt
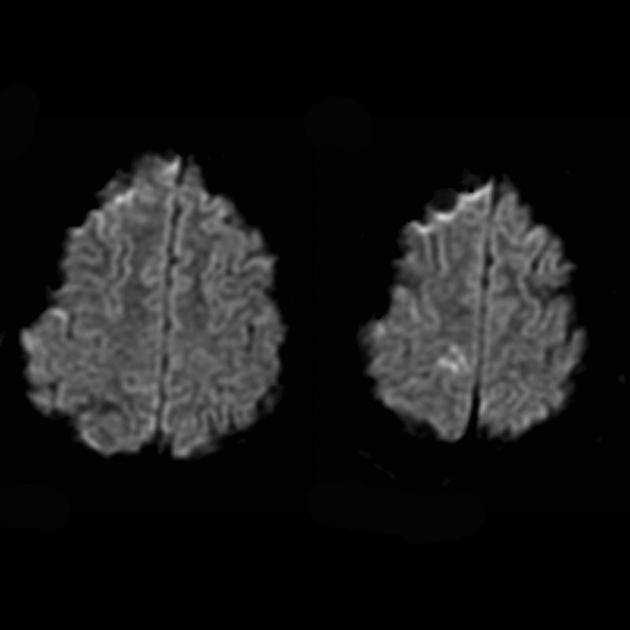
För differentiering med stroke används ADC och DWI (diffusionsrestriktion vid stroke (cytotoxiskt ödem), och vid MELAS är diffusionen något begränsad eller oförändrad (vasogent ödem).
Engagemang i den patologiska processen av den subkortikala vita substansen i hjärnan.
Försämring av visualiseringen av klarheten i konturerna av veckningarna och en ökning av signalen från dem på T2-viktade bilder.
Kronisk infarkt
Förändringar kan vara symmetriska eller asymmetriska.
Fokal atrofi uppstår mot bakgrund av regression och klinisk förbättring.
Hjärnans parietallober, occipitallober och temporallober är vanligast drabbade.
MR-spektroskopi
Ökade laktatnivåer.




Materialet är avsett för neurologer, terapeuter och allmänläkare.
Sergey Likhachev, chef, MD. vetenskaper, professor;
Inessa Pleshko, ledande forskare, Ph.D. Vetenskaper, Neurologiska avdelningen vid det republikanska vetenskapliga och praktiska centret för neurologi och neurokirurgi.
Cerebral autosomal dominant arteriopati med subkortikala infarkter och leukoencefalopati (CADASIL) är en progressiv autosomal dominant sjukdom, vars kliniska manifestationer inkluderar återkommande subkortikala ischemiska stroke, migrän, subkortikal demens och affektiva störningar. Nuvarande prevalens - 1 fall
per 100 000 invånare.
Det republikanska vetenskapliga och praktiska centret för neurologi och neurokirurgi ser 7 patienter (inklusive 4 kvinnor) med CADASIL; ålder - från 32 till 68 år. De undersöktes med neurologiska, molekylärgenetiska metoder. Det fanns karakteristiska symtom; i historien - migrän, återkommande lakunära stroke och affektiva störningar. Hjärn-MR avslöjade subkortikala infarkter och leukoencefalopati karakteristiskt för CADASIL.
Som ett resultat av molekylärgenetisk diagnostik hade 2 personer en heterozygot mutation i Notch3-genen på den 19:e kromosomen, vilket orsakar CADASIL. Notch-gener kodar för transmembranreceptorer involverade i cellontogeni. Med CADASIL bestäms i de flesta fall missense-mutationer, på grund av vilka strukturen hos transmembranproteinet förändras och dess funktioner försämras.
Patogenesen för CADASIL är inte helt klar. Man tror att huvudfaktorn är arteriopati med progressiv ocklusion av små perforerande kärl i hjärnans vita substans (som leder till kronisk hypoperfusion). Samtidigt hittas karakteristiska granulära osmiofila inneslutningar, vilket orsakar proliferation av basalmembrankomponenter, förtjockning av mittmembranet och mekanisk kompression av små artärer. Som ett resultat skadas blod-hjärnbarriären - ödem utvecklas.
En ytterligare patologisk faktor är aktiveringen av astrocyter nära kärlväggen. De frisätter endotel-1, vilket orsakar vasokonstriktion och försämrat blodflöde.
Sammansättningen av granulära osmiofila inneslutningar är okänd. Det antas att Notch3-proteinet är en av deras komponenter. I hudbiopsier av patienter med en Notch3-mutation kan osmiofila granuler och degeneration av glatta muskelceller detekteras redan före 20 års ålder.
Klinisk diagnostik av CADASIL:
- belastad familjehistoria;
- utvecklingen av de första symptomen på sjukdomen före 50 års ålder;
- förekomsten av två av följande symtom - migrän, återkommande stroke, humörstörningar, subkortikal demens.
Vaskulära riskfaktorer som är etiologiskt associerade med neurologiska symtom bör uteslutas. MRT visar skador på den vita substansen i hjärnhalvorna och frånvaron av kortikala infarkter.
En tillförlitlig diagnos av "CADASIL" bekräftas av ett positivt resultat av molekylärgenetisk diagnos eller upptäckt av arteriopati med karakteristiska granulära osmiofila inneslutningar i hud- eller muskelbiopsi.
De vanligaste symtomen på CADASIL är övergående ischemiska attacker och ischemiska stroke, observerade hos nästan 85 % av patienterna.
De kännetecknas av ett återkommande förlopp, manifesterat av klassiska syndrom av lakunära stroke och fullständig klinisk remission efter några dagar eller veckor.
Den näst vanligaste är kognitiva funktionsnedsättningar (noteras hos 60 % av patienterna). Kan börja vid 35 års ålder, ibland till och med före ischemiska episoder. Cirka 75 % av CADASIL-patienterna utvecklar demens. Det första symtomet är vanligtvis migrän; inträffar ofta före 20 års ålder och föregår vanligtvis stroke.
Data om hjärtats inblandning i den patologiska processen i CADASIL är motsägelsefulla. L. Oberstein et al. (2003) fann att 25 % av patienterna med diagnosen CADASIL hade en historia av akut hjärtinfarkt eller Q-vågspatologi på elektrokardiogrammet. I en annan studie visade Cumurciuc et al. (2006) fann ingen positiv hjärthistoria hos 23 personer med en Notch3-mutation.
Kliniska manifestationer av CADASIL och cerebral mikroangiopati av en annan etiologi är likartade - differentialdiagnos krävs.
För att i tid bestämma CADASIL hos patienter och deras familjer är det nödvändigt att tillgripa molekylärgenetiska metoder och / eller histologiska studier.
MELAS syndrom
Mitokondriell encefalomyopati med laktacidos och strokeliknande episoder (MELAS) är en sällsynt ärftlig sjukdom som orsakas av patologi i mitokondriella genom, försämrad energimetabolism och funktion av de mest energiberoende organen och vävnaderna (CNS, hjärt- och skelettmuskler, ögon, njurar, lever, benmärg, endokrina systemet). Den stora variationen av kliniska manifestationer av MELAS-syndromet och den sällsynta förekomsten förutbestämmer diagnostiska svårigheter för läkaren.
I det republikanska vetenskapliga och praktiska centret för neurologi och neurokirurgi observeras 3 patienter (en 46-årig kvinna och hennes söner i åldrarna 24 och 23) med ett diagnostiserat MELAS-syndrom. De genomgick klinisk och neurologisk undersökning, molekylärgenetisk diagnostik, MR av hjärnan.
Alla är korta; i historien - symtom på mitokondriell patologi: sensorineural hörselnedsättning, migränliknande huvudvärk, dålig träningstolerans. Sjukdomens debut är generaliserade konvulsiva anfall. Hos 2 patienter uppträdde de första symtomen före 20 års ålder; det förekom epileptiska anfall som följde efter varandra, episoder av synnedsättning med närvaro av foci i neuroimaging i de occipital- och temporala regionerna, en ökning av laktatnivån i blodet och cerebrospinalvätskan. 1 person hade en måttlig minskning av kognitiva funktioner; enligt ultraljud av hjärtat - hypertrofisk kardiomyopati; diabetes.
En molekylärgenetisk studie avslöjade multisystemlesioner typiska för MELAS, stor variation och varierande grad av kliniska manifestationer, motsvarande antalet mutantkopior av A3243G i tRNA Leu(UUR)-genen.
MELAS kännetecknas av en moderlig typ av arv, förekomsten av sporadiska fall när en de novo mutation inträffar; ackumulering i celler - både normala och mutanta typer - av mitokondriellt DNA (heteroplasmi) och slumpmässig fördelning under delning mellan dotterceller (mitotisk segregation). På den genetiska nivån är orsaken till MELAS-syndrom den heteroplasmatiska omarrangemanget 3243A>G i tRNALeu(UUR)-genen (80 % av fallen detekteras).
Sjukdomens patogenes har ännu inte studerats. Det finns 2 huvudteorier - "mitokondriell angiopati" och "mitokondriell cytopati". Det är känt att den strokeliknande lesionen inte motsvarar de vaskulära zonerna och sträcker sig till de omgivande områdena på grund av samtidigt vasogent ödem orsakat av förlängd epileptisk aktivitet. Som föreslagits beror strokeliknande episoder på neural hyperexcitabilitet i ett begränsat område av hjärnan. Det uppstår från mitokondriell dysfunktion i kapillära endotelceller eller i neuroner eller i astrocyter; depolariserar intilliggande neuroner, vilket leder till spridning av epileptisk aktivitet.
Dessutom har patienter med MELAS i intervallen mellan strokeliknande episoder, enligt singelfoton emission computed tomography (SPECT), hypoperfusion av den bakre cingulate cortex, vilket indikerar en störning av cerebral hemodynamik.
Brott mot oxidativ fosforylering, bristning av den mitokondriella andningskedjan bidrar till dominansen av katabolisk metabolism och förändringar från Krebs-cykeln till anaerob glykos med laktatackumulering. En hög nivå av det senare i CNS korrelerar vanligtvis med perioder av neurologiska symtom.
De huvudsakliga kliniska tecknen på MELAS är strokeliknande episoder, laktacidos och närvaron av "slitna röda fibrer" i muskelbiopsiprover. Ytterligare manifestationer kan vara demens, psykos, epileptiska anfall, migränliknande huvudvärk, ataxi, myopati, förkalkning av basalganglierna vid neuroimaging, optisk atrofi, retinopati, dövhet, diabetes, intestinal pseudo-obstruktion, kardiomyopati.
Den tidiga åldern för MELAS debut är från 5 till 20 år, men det finns observationer av en sen debut - i 5:e–6:e decennierna av livet. Det finns fall när syndromet började efter hjärtsjukdomar.
Den multisystemiska karaktären hos lesioner i MELAS komplicerar klinisk diagnos.
Sjukdomens ärftliga natur tvingar att genomföra molekylärgenetiska studier för att göra en korrekt diagnos.
och identifiera andra patienter - bland anhöriga till patienten.
Materialet är avsett för neurologer, terapeuter och allmänläkare.
| Mitokondriell myopati, encefalomyopati, laktacidos och strokeliknande episoder | |
|---|---|
| Basalgangliaförkalkning, cerebellär atrofi, ökat laktat; CT-bild av en person med diagnosen MELAS | |
| Specialitet | neurologi |
genetik
Muskelbiopsi av en person som diagnostiserats med MELAS, men som inte bär på en känd mutation. (a) Gomoris modifierade trefärgade färgämne visar några trasiga röda fibrer (pilar). (b) Cytokrom c-oxidasfärgning, som visar typ-1, lätt färgade och typ II-fibrer, mörka fibrer och några fibrer med onormala mitokondriella samlingar (pilar). Observera att cytokrom c-oxidasnegativa fibrer vanligtvis ses vid mitokondriell encefalopati, laktacidos och strokeliknande episoder (MELAS). (c) Succinatdehydrogenasfärgning visar flera trasiga blå fibrer och intensiv färgning i blodkärlsmitokondrier (pil). (d) Elektronmikroskopi visar en onormal samling av mitokondrier med parakristallina inneslutningar (pilar), osmiofila inneslutningar (stora pilar) och mitokondriella vakuoler (små pilar).
MELAS orsakas av mutationer i gener i mitokondrie-DNA.
NADH-dehydrogenaser
Mutationer i MT-TL1 orsaka över 80 procent av alla MELAS-fall. De minskar mitokondriernas förmåga att tillverka proteiner, använda syre och producera energi. Forskare har inte bestämt hur förändringar i mitokondrie-DNA leder till de specifika tecknen och symtomen på MELAS. De fortsätter att undersöka effekterna av mitokondriella genmutationer i olika vävnader, särskilt i hjärnan.
arv
Detta tillstånd ärvs i ett mitokondriellt mönster, som också är känt som moderns arv och heteroplasmi. Detta arvsmönster hänvisar till gener som finns i mitokondrie-DNA. Eftersom ägg, men inte spermier, bidrar med mitokondrier till det utvecklande embryot, är det bara honor som går igenom mitokondriella tillstånd för sina barn. Mitokondriella störningar kan förekomma i varje generation av en familj och kan drabba både män och kvinnor, men fäder för inte över mitokondriella egenskaper hos sina barn. I de flesta fall ärver personer med MELAS den förändrade mitokondriella genen från sin mamma. Mindre vanligt är sjukdomen ett resultat av en ny mutation i den mitokondriella genen och förekommer hos personer utan en familjehistoria av MELAS.
diagnostik
Behandling/prognos
Patienter hanteras efter vilka delar av kroppen som påverkas vid en viss tidpunkt.